General Information About Retinoblastoma
Retinoblastoma is a pediatric cancer that requires careful integration of multidisciplinary care. Treatment of retinoblastoma aims to save the patient's life and preserve useful vision. For patients presenting with extraocular retinoblastoma, treatment with systemic chemotherapy and radiation therapy is likely to be curative. However, extraorbital disease requires intensive chemotherapy and may include consolidation with high-dose chemotherapy and autologous hematopoietic stem cell rescue with or without radiation therapy. While a large proportion of patients with systemic extra–central nervous system (CNS) metastases can be cured, the prognosis for patients with intracranial disease is dismal.
Incidence
Retinoblastoma is a relatively uncommon tumor of childhood that arises in the retina. It accounts for about 3% of the cancers occurring in children younger than 15 years.
Retinoblastoma is a cancer of the very young child. Two-thirds of all cases of retinoblastoma are diagnosed before age 2 years.[1] Thus, while the estimated annual incidence in the United States is approximately 3 cases per 1 million children younger than 20 years, the age-adjusted annual incidence in children aged 0 to 4 years is 18.4 cases per 1 million.[2]
Anatomy
Retinoblastoma arises in the retina, and it may grow under the retina and/or toward the vitreous cavity. Involvement of the ocular coats and optic nerve occurs as a sequence of events as the tumor progresses.
Focal invasion of the choroid is common, although massive invasion occurs in cases of advanced disease. After invading the choroid, the tumor gains access to systemic circulation and creates the potential for metastases. Further progression through the ocular coats leads to invasion of the sclera and the orbit. Tumors that invade the anterior chamber may gain access to systemic circulation through the canal of Schlemm. Progression through the optic nerve and past the lamina cribrosa increases the risk of systemic and CNS dissemination (see Figure 1).
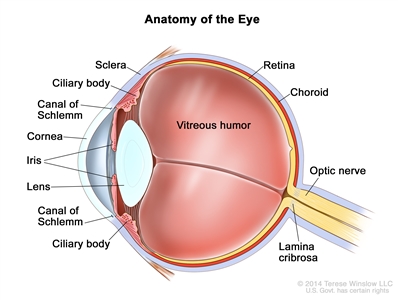
Figure 1. Anatomy of the eye showing the sclera, ciliary body, canal of Schlemm, cornea, iris, lens, vitreous humor, retina, choroid, optic nerve, and lamina cribrosa. The vitreous humor is a gel that fills the center of the eye.
Screening
Consensus reports from the American Association of Ophthalmic Oncologists and Pathologists and the American Association for Cancer Research Childhood Cancer Predisposition Workshop describe screening guidelines for children at risk of developing retinoblastoma.[3,4]
In children with a positive family history of retinoblastoma, early-in-life screening by fundus examination is performed under general anesthesia at regular intervals. Examinations are performed according to a schedule based on the absolute estimated risk, as determined by identification of the RB1 variant in the family and the presence of the RB1 variant in the child.[3,4]
Infants born to affected parents have a dilated eye examination under anesthesia as soon as possible in the first month of life, and a genetic evaluation is performed. Infants with a positive genetic test are examined under anesthesia on a monthly basis. In infants who do not develop disease, monthly examinations continue throughout the first year. The frequency of those examinations may be decreased progressively during the second and subsequent years. Screening children with a positive family history of retinoblastoma can improve their prognosis, in terms of globe sparing and use of less intensive, ocular-salvage treatments (see Table 1 and Figure 2).[3,4]
Table 1. Pretest Risk for Relatives to Carry the MutantRB1Allele of the Probanda,b
| Relative of Proband |
Pretest Risk for Mutant Allele (%) |
|
|
Bilateral Proband (100) |
Unilateral Proband (15) |
|
a Reprinted fromOphthalmology, Volume 125, Issue 3, Alison H. Skalet, Dan S. Gombos, Brenda L. Gallie, Jonathan W. Kim, Carol L. Shields, Brian P. Marr, Sharon E. Plon, Patricia Chévez-Barrios, Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists, Pages 453–458, Copyright (2018), with permission from Elsevier. |
|
b Pretest risk forRB1mutation in family members of an affected child withretinoblastoma. Risk forRB1mutant allele is shown as a percentage for unilateral and bilateral probands without family history of retinoblastoma. |
|
c Third- and fourth-degree relatives of unilateral probands have calculated risks of 0.003% and 0.001%, respectively, which are less than the normal population risk of 0.007% (1 in 15,000 live births); therefore, the risk is stated at 0.007%. |
| Offspring (infant) |
50 |
7.5 |
| Parent |
5 |
0.8 |
| Sibling |
2.5 |
0.4 |
| Niece/nephew |
1.3 |
0.2 |
| Aunt/uncle |
0.1 |
0.007c |
| First cousin |
0.05 |
0.007c |
| General population |
0.007 |
|
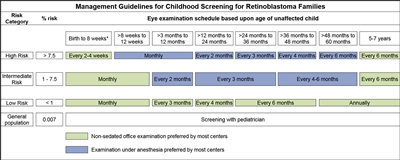
Figure 2. Management guidelines for childhood screening for retinoblastoma. The presented schedules are general guidelines and reflect a schedule for examinations in which no lesions of concern are noted. It may be appropriate to examine some children more frequently. Decisions regarding examination method, examination under anesthesia (EUA) versus nonsedated examination in the office, are complex and best decided by the clinician in discussion with the patient's family. The preference of the majority of the clinical centers involved in the creation of this consensus statement is reflected, but individual centers may make policy decisions based on available resources and expert clinician preference. Examination under anesthesia will be strongly considered for any child who is unable to participate in an office examination sufficiently to allow thorough examination of the retina. *A minority of clinical centers also prefer EUA for high- and intermediate-risk children (calculated risk >1%) from birth to 8 weeks of age. Reprinted from Ophthalmology, Volume 125, Issue 3, Alison H. Skalet, Dan S. Gombos, Brenda L. Gallie, Jonathan W. Kim, Carol L. Shields, Brian P. Marr, Sharon E. Plon, Patricia Chévez-Barrios, Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists, Pages 453–458, Copyright (2018), with permission from Elsevier.
It is common practice to use ophthalmic examinations to screen the parents and siblings of patients with retinoblastoma to exclude an unknown familial disease. However, in the absence of genetic testing, the screening plan for a child with a biological parent who had unilateral retinoblastoma is not well defined.[5]
Clinical Presentation
Age at presentation correlates with laterality. Patients with bilateral disease present at a younger age, usually in the first 12 months of life.
Most patients present with leukocoria, which is occasionally first noticed after a flash photograph is taken. Strabismus is the second most common presenting sign and usually correlates with macular involvement. Very advanced intraocular tumors present with pain, orbital cellulitis, glaucoma, or buphthalmos.
As the tumor progresses, patients may present with orbital or metastatic disease. Metastases occur in the preauricular and laterocervical lymph nodes, in the CNS, or systemically (commonly in the bones, bone marrow, and liver).
In the United States, Hispanic children and children living in lower socioeconomic conditions have presented with more advanced disease.[6]
Diagnostic and Staging Evaluation
Diagnostic evaluation of retinoblastoma includes the following:
-
Eye examination. Intraocular retinoblastoma is usually diagnosed without pathological confirmation. An examination under anesthesia with a maximally dilated pupil and scleral indentation is required to inspect the entire retina. The number, location, and size of tumors; the presence of retinal detachment and subretinal fluid; and the presence of subretinal and vitreous seeds must be documented in detail.
-
Ocular ultrasonography and magnetic resonance imaging (MRI). Bidimensional ocular ultrasonography and MRI can be useful to differentiate retinoblastoma from other causes of leukocoria and in the evaluation of extrascleral and extraocular extension in children with advanced intraocular retinoblastoma. Optic nerve enhancement by MRI does not necessarily indicate involvement. Cautious interpretation of those findings is needed.[7]
Patients with suspected extraocular extension by imaging or high-risk pathology in the enucleated eye (i.e., massive choroidal invasion or involvement of the sclera or the optic nerve beyond the lamina cribrosa) may need to be evaluated for the presence of metastatic disease. Patients presenting with these pathological features in the enucleated eye are at high risk of developing metastases. In these cases, the following procedures may be performed:[8]
- Bone scintigraphy.
- Bone marrow aspiration and biopsy.
- Lumbar puncture.
Genetics and Genomics of Retinoblastoma
Retinoblastoma is a tumor that occurs in heritable (25%–30%) and nonheritable (70%–75%) forms.
Heritable Retinoblastoma
Heritable retinoblastoma is defined by the presence of a germline pathogenic variant of the RB1 gene. This germline pathogenic variant may have been inherited from an affected progenitor (25% of cases) or may have occurred in a germ cell before conception or in utero during early embryogenesis in patients with sporadic disease (75% of cases). The presence of positive family history or bilateral or multifocal disease is suggestive of heritable disease.
Heritable retinoblastoma may manifest as unilateral or bilateral disease. The penetrance of the RB1 variant (laterality, age at diagnosis, and number of tumors) is probably dependent on concurrent genetic modifiers such as MDM2 and MDM4 polymorphisms.[9,10] All children with bilateral disease and approximately 15% of patients with unilateral disease are presumed to have the heritable form, even though only 25% have an affected parent. In a series of 482 patients with unilateral retinoblastoma, germline pathogenic variants were identified in 33% of infants younger than 12 months, 6% of children aged 12 to 24 months, and 7% of children aged 24 to 39 months. The highest incidence of germline retinoblastoma was in patients younger than 1 year compared with patients older than 1 year (odds ratio, 2.96).[11][Level of evidence C2]
Children with heritable retinoblastoma tend to be diagnosed at a younger age than children with the nonheritable form of the disease.[12]
Nonheritable Retinoblastoma
The genomic landscape of retinoblastoma is driven by alterations in RB1 that lead to biallelic inactivation.[13,14] A rare cause of RB1 inactivation is chromothripsis, which may be difficult to detect by conventional methods.[15]
Recurrent changes in genes other than RB1 are uncommon in retinoblastoma but do occur. Variants or deletions of BCOR and amplification of MYCN are the most frequently reported events.[13,14,15,16,17,18] A study of 1,068 unilateral nonfamilial retinoblastoma tumors reported that 2% to 3% of tumors lacked evidence of RB1 loss and approximately one-half of these cases without evidence of RB1 loss showed MYCN amplification.[14] However, MYCN amplification is also observed in retinoblastoma tumors that have RB1 alterations, suggesting that inactivation of RB1 by a variant or an inactive retinoblastoma protein is a requirement for the development of retinoblastoma, independent of MYCN amplification.[19]
Genetic Counseling
Genetic counseling is an integral part of the management of patients with retinoblastoma and their families, regardless of clinical presentation. Counseling includes a discussion of the main forms of retinoblastoma, which helps parents understand the genetic consequences of each form of retinoblastoma and estimate the risk of disease in family members.[20] Counseling also includes guidance toward appropriate screening for both patients and their families, especially if the risk of developing a second primary malignancy is increased.
Genetic counseling, however, is not always straightforward. Approximately 10% of children with retinoblastoma have somatic genetic mosaicism, which contributes to the difficulty of genetic counseling.[21] Children with mosaic alleles have fewer tumors, and the tumors are more likely to remain unilateral.[22] In addition, for one specific variant, the risk of retinoblastoma in a sibling may depend partly on whether the variant is inherited from the mother or father.[23] For more information, see Cancer Genetics Risk Assessment and Counseling.
Genetic Testing
Blood and tumor samples can be tested to determine whether a patient with retinoblastoma has a germline or somatic variant in the RB1 gene. Once the patient's genetic variant has been identified, other family members can be screened directly for the variant with targeted sequencing.
A multistep assay that includes the following may be performed for a complete genetic evaluation of the RB1 gene:[24]
- DNA sequencing to identify variants within coding exons and immediate flanking intronic regions plus the promoter regions.
- Duplication/deletion analysis.
- Methylation analysis of the RB1 promoter region on DNA isolated from the tumor.
In cases of somatic mosaicism or cytogenetic abnormalities, the variants may not be easily detected. More exhaustive techniques such as karyotyping, fluorescence in situ hybridization, and methylation analysis of the RB1 promoter may be needed. Deep (2500x) sequencing of an RB1 genomic amplicon from lymphocyte DNA can reveal low-level mosaicism.[25] Because mosaicism is caused by a postzygotic variant, such a finding obviates the need for serial examination of siblings under anesthesia. Current technologies will not discover some mosaic variants at very low levels of amplification, variants outside of the RB1 coding exons or the flanking intronic regions, variants not found in lymphocytes but in other tissues (mosaic), or mosaic large rearrangements of RB1.[25] Combining the above techniques, a germline pathogenic variant may be detected in more than 90% of patients with heritable retinoblastoma.[20,26,27]
The absence of detectable somatic RB1 variants in approximately 3% of unilateral, nonheritable retinoblastoma cases suggests that alternative genetic mechanisms may underlie the development of retinoblastoma.[28] In one-half of these cases, high levels of MYCN amplification have been reported. These patients had distinct, aggressive histological features and a median age at diagnosis of 4 months.[14] However, MYCN amplification has also been reported to coexist with RB1 variants.[19] In another small subset of tumors without detectable somatic RB1 variants, chromothripsis is responsible for inactivating the RB1 gene.[15]
Postdiagnosis Surveillance
Children with a germline RB1 pathogenic variant may continue to develop new tumors for a few years after diagnosis and treatment. For this reason, these patients need to be examined frequently. It is common practice for examinations to occur every 2 to 4 months for at least 28 months.[29] The interval between examinations is based on the stability of the disease and age of the child (i.e., less frequent visits as the child ages).
A proportion of children who present with unilateral retinoblastoma will eventually develop disease in the opposite eye. Periodic examinations of the unaffected eye are performed until the germline status of the RB1 gene is determined.
Because of the poor prognosis for patients with trilateral retinoblastoma, screening with neuroimaging until age 5 years is a common practice in the monitoring of children with the heritable form of the disease. For more information, see the Trilateral retinoblastoma section.
Causes of Retinoblastoma-Related Mortality
While retinoblastoma is a highly curable disease, the challenge is to preserve life and to prevent the loss of an eye, blindness, and other serious effects of treatment that reduce the patient's life span or quality of life. With improvements in the diagnosis and management of retinoblastoma over the past several decades, metastatic retinoblastoma is observed less frequently in the United States and other developed nations. As a result, other causes, such as trilateral retinoblastoma and subsequent neoplasms (SNs), have become significant contributors to retinoblastoma-related mortality in the first and subsequent decades of life. In the United States, before the advent of chemoreduction as a means of treating heritable or bilateral disease and the implementation of neuroimaging screening, trilateral retinoblastoma contributed to more than 50% of retinoblastoma-related mortality for patients in the first decade after their diagnosis.[30] For more information about SNs, see the Late Effects of Retinoblastoma Therapy section.
Trilateral retinoblastoma
Trilateral retinoblastoma is a well-recognized syndrome that occurs in 5% to 15% of patients with heritable retinoblastoma. It is defined by the development of an asynchronous intracranial midline neuroblastic tumor, which typically develops between the ages of 20 and 36 months.[31]
Because of the poor prognosis and the apparent improved survival with early detection and aggressive treatment of trilateral retinoblastoma, screening with routine neuroimaging could potentially detect most cases within 2 years of the first retinoblastoma diagnosis.[31] Routine baseline brain MRI is recommended at diagnosis because it may detect trilateral retinoblastoma at a subclinical stage. In a small series, the 5-year overall survival rate was 67% for patients with tumors that were detected at baseline, compared with 11% for the group with a delayed diagnosis.[32]
Although it is not clear whether early diagnosis can impact survival, screening with MRI has been recommended as often as every 6 months for 5 years for patients suspected of having heritable disease or those with unilateral disease and a positive family history.[33] Computed tomography scans are generally avoided for routine screening in these children because of the risk related to ionizing radiation exposure.
A cystic pineal gland, which is commonly detected by surveillance MRI, needs to be distinguished from a cystic variant of pineoblastoma. In children without retinoblastoma, the incidence of pineal cysts has been reported to be 55.8%.[34] In a case-control study that included 77 children with retinoblastoma and 77 controls, the incidence of pineal cysts was similar (61% and 69%, respectively), and the size and volume of the pineal gland was not significantly different between the groups.[35] However, a cystic component has been described in up to 57% of patients with histologically confirmed trilateral retinoblastoma.[32] An excessive increase in the size of the pineal gland seems to be the strongest parameter indicating a malignant process.[35]
References:
-
Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed December 22, 2023.
-
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
-
Skalet AH, Gombos DS, Gallie BL, et al.: Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology 125 (3): 453-458, 2018.
-
Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
-
Abramson DH: Re: Skalet et al.: Screening children at risk for retinoblastoma: consensus report from the American Association of Ophthalmic Oncologists and Pathologists (Ophthalmology. 2018;125:453-458). Ophthalmology 125 (9): e63-e64, 2018.
-
Truong B, Green AL, Friedrich P, et al.: Ethnic, Racial, and Socioeconomic Disparities in Retinoblastoma. JAMA Pediatr 169 (12): 1096-104, 2015.
-
Khurana A, Eisenhut CA, Wan W, et al.: Comparison of the diagnostic value of MR imaging and ophthalmoscopy for the staging of retinoblastoma. Eur Radiol 23 (5): 1271-80, 2013.
-
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
-
Castéra L, Sabbagh A, Dehainault C, et al.: MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst 102 (23): 1805-8, 2010.
-
de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, et al.: Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer 59 (1): 39-43, 2012.
-
Shields CL, Dockery P, Ruben M, et al.: Likelihood of Germline Mutation With Solitary Unilateral Retinoblastoma Based on Patient Age at Presentation: Analysis of 482 Consecutive Patients. J Pediatr Ophthalmol Strabismus 58 (6): 355-364, 2021 Nov-Dec.
-
Andreoli MT, Chau FY, Shapiro MJ, et al.: Epidemiological trends in 1452 cases of retinoblastoma from the Surveillance, Epidemiology, and End Results (SEER) registry. Can J Ophthalmol 52 (6): 592-598, 2017.
-
Zhang J, Benavente CA, McEvoy J, et al.: A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481 (7381): 329-34, 2012.
-
Rushlow DE, Mol BM, Kennett JY, et al.: Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14 (4): 327-34, 2013.
-
McEvoy J, Nagahawatte P, Finkelstein D, et al.: RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5 (2): 438-50, 2014.
-
Afshar AR, Pekmezci M, Bloomer MM, et al.: Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 127 (6): 804-813, 2020.
-
Kooi IE, Mol BM, Massink MP, et al.: Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6: 25264, 2016.
-
Francis JH, Richards AL, Mandelker DL, et al.: Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers (Basel) 13 (1): , 2021.
-
Ewens KG, Bhatti TR, Moran KA, et al.: Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med 6 (3): 619-630, 2017.
-
Richter S, Vandezande K, Chen N, et al.: Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet 72 (2): 253-69, 2003.
-
Dommering CJ, Mol BM, Moll AC, et al.: RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet 51 (6): 366-74, 2014.
-
Reddy MA, Butt M, Hinds AM, et al.: Prognostic Information for Known Genetic Carriers of RB1 Pathogenic Variants (Germline and Mosaic). Ophthalmol Retina 5 (4): 381-387, 2021.
-
Eloy P, Dehainault C, Sefta M, et al.: A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genet 12 (2): e1005888, 2016.
-
Clark R: Retinoblastoma: genetic testing and counseling. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 441-6.
-
Amitrano S, Marozza A, Somma S, et al.: Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet 23 (11): 1523-30, 2015.
-
Sagi M, Frenkel A, Eilat A, et al.: Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer 14 (3): 471-80, 2015.
-
Chen Z, Moran K, Richards-Yutz J, et al.: Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat 35 (3): 384-91, 2014.
-
Nichols KE, Houseknecht MD, Godmilow L, et al.: Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat 25 (6): 566-74, 2005.
-
Abramson DH, Mendelsohn ME, Servodidio CA, et al.: Familial retinoblastoma: where and when? Acta Ophthalmol Scand 76 (3): 334-8, 1998.
-
Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
-
de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
-
Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
-
Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
-
Sirin S, de Jong MC, Galluzzi P, et al.: MRI-based assessment of the pineal gland in a large population of children aged 0-5 years and comparison with pineoblastoma: part II, the cystic gland. Neuroradiology 58 (7): 713-21, 2016.
-
Pham TT, Siebert E, Asbach P, et al.: Magnetic resonance imaging based morphologic evaluation of the pineal gland for suspected pineoblastoma in retinoblastoma patients and age-matched controls. J Neurol Sci 359 (1-2): 185-92, 2015.
Tumor Pathology of Retinoblastoma
Maturing cone precursor cells appear to be the cell of origin in human retinoblastoma.[1,2] Microscopically, the appearance of retinoblastoma depends on the degree of differentiation. Undifferentiated retinoblastoma is composed of small, round, densely packed cells with hypochromatic nuclei and scant cytoplasm. Several degrees of photoreceptor differentiation have been described and are characterized by distinctive arrangements of tumor cells, as follows:
-
Flexner-Wintersteiner rosettes are specific to retinoblastoma. These structures consist of a cluster of low columnar cells arranged around a central lumen that is bounded by an eosinophilic membrane analogous to the external membrane of the normal retina. The lumen contains rosettes that are seen in 70% of tumors.
-
Homer Wright rosettes are composed of irregular circlets of tumor cells arranged around a tangle of fibrils with no lumen or internal-limiting membrane. Homer Wright rosettes are infrequently seen in retinoblastoma and are most often seen in other neuroblastic tumors, such as neuroblastoma and medulloblastoma.
Retinoblastomas are characterized by marked cell proliferation, as evidenced by high mitosis counts, extremely high MIB-1 labeling indices, and strong diffuse nuclear immunoreactivity for CRX, a useful marker to discriminate retinoblastoma from other malignant, small, round cell tumors.[3,4]
Cavitary retinoblastoma, a rare variant of retinoblastoma, has ophthalmoscopically visible lucent cavities within the tumor. The cavitary spaces appear hollow on ultrasonography and hypofluorescent on angiography. Histopathologically, the cavitary spaces have been shown to represent areas of photoreceptor differentiation.[5]
Cavitary retinoblastoma has been associated with minimal visible response to intravenous and intra-arterial chemotherapy, which is thought to be a sign of tumor differentiation.[6,7] Despite the blunted clinical response, patients with cavitary retinoblastoma have favorable long-term outcomes, with good tumor response and globe salvage that is similar to that in patients with noncavitary retinoblastoma.
A pathologist experienced in ocular pathology and retinoblastoma should examine the enucleated specimen, particularly to determine risk features of extraocular dissemination. For more information, see the Treatment of Intraocular Retinoblastoma section.
References:
-
Xu XL, Singh HP, Wang L, et al.: Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 514 (7522): 385-8, 2014.
-
Singh HP, Wang S, Stachelek K, et al.: Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc Natl Acad Sci U S A 115 (40): E9391-E9400, 2018.
-
Terry J, Calicchio ML, Rodriguez-Galindo C, et al.: Immunohistochemical expression of CRX in extracranial malignant small round cell tumors. Am J Surg Pathol 36 (8): 1165-9, 2012.
-
Schwimer CJ, Prayson RA: Clinicopathologic study of retinoblastoma including MIB-1, p53, and CD99 immunohistochemistry. Ann Diagn Pathol 5 (3): 148-54, 2001.
-
Palamar M, Pirondini C, Shields CL, et al.: Cavitary retinoblastoma: ultrasonographic and fluorescein angiographic findings in 3 cases. Arch Ophthalmol 126 (11): 1598-600, 2008.
-
Mashayekhi A, Shields CL, Eagle RC, et al.: Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology 112 (6): 1145-50, 2005.
-
Rojanaporn D, Kaliki S, Bianciotto CG, et al.: Intravenous chemoreduction or intra-arterial chemotherapy for cavitary retinoblastoma: long-term results. Arch Ophthalmol 130 (5): 585-90, 2012.
Staging and Grouping Systems for Retinoblastoma
The staging of patients with retinoblastoma requires close coordination of radiologists, pediatric oncologists, and ophthalmologists. Several staging and grouping systems have been proposed for retinoblastoma.[1] Overall assessment of retinoblastoma extension is documented by staging systems. The extent of intraocular disease, which is relevant for ocular salvage, is documented by grouping systems. For treatment purposes, retinoblastoma is categorized into intraocular and extraocular disease.
Intraocular Retinoblastoma
Intraocular retinoblastoma is localized to the eye. It may be confined to the retina or may extend to involve other structures such as the choroid, ciliary body, anterior chamber, and optic nerve head. Intraocular retinoblastoma, however, does not extend beyond the eye into the tissues around the eye or to other parts of the body.
Extraocular Retinoblastoma
Extraocular retinoblastoma extends beyond the eye. It may be confined to the tissues around the eye (orbital retinoblastoma), it may have spread to the central nervous system, or it may have spread systemically to the bone marrow or lymph nodes (metastatic retinoblastoma).
Staging Systems
American Joint Committee on Cancer (AJCC) staging system
Several staging systems have been proposed over the years. The newest standard for state-mandated cancer reporting to the North American Association of Cancer Registries requires AJCC staging, according to the 8th edition of the staging manual.[2]
For information about the clinical classification definitions of primary tumor (T), regional lymph node (N), distant metastasis (M), histological grade, and prognostic stage groups, see Table 3, Table 5, Table 7, Table 8, and Table 9.
For information about the pathological classification definitions of T, N, M, histological grade, and prognostic stage groups, see Table 4, Table 6, Table 7, Table 8, and Table 10.
This staging system affects cases diagnosed in 2018 and thereafter. Retinoblastoma staging is the first to acknowledge the role of genetic predisposition by incorporating an H category. H1 refers to patients with bilateral or trilateral retinoblastoma, a family history of retinoblastoma, or the presence of an RB1 variant (see Table 2).[2]
Table 2. Definition of Heritable Trait (H)a
| H Category |
H Criteria |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| HX |
Unknown or insufficient evidence of a constitutionalRB1gene variant |
| H0 |
NormalRB1alleles in blood tested with demonstrated high-sensitivity assays |
| H1 |
Bilateral retinoblastoma, retinoblastoma with an intracranial primitive neuroectodermal tumor (i.e., trilateral retinoblastoma), patient with family history of retinoblastoma,or molecular definition of a constitutionalRB1gene variant |
Table 3. Definition of Clinical Primary Tumor (cT)a
| cT Category |
cT Criteria |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| cTX |
Unknown evidence of intraocular tumor |
| cT0 |
No evidence of intraocular tumor |
| cT1 |
Intraretinal tumor(s) with subretinal fluid ≤5 mm from the base of any tumor |
| |
cT1a |
Tumors ≤3 mm and further than 1.5 mm from disc and fovea |
| |
cT1b |
Tumors >3 mm or closer than 1.5 mm from disc or fovea |
| cT2 |
Intraocular tumor(s) with retinal detachment, vitreous seeding, or subretinal seeding |
| |
cT2a |
Subretinal fluid >5 mm from the base of any tumor |
| |
cT2b |
Vitreous seeding and/or subretinal seeding |
| cT3 |
Advanced intraocular tumor(s) |
| |
cT3a |
Phthisis or pre-phthisis bulbi |
| |
cT3b |
Tumor invasion of choroid, pars plana, ciliary body, lens, zonules, iris, or anterior chamber |
| |
cT3c |
Raised intraocular pressure with neovascularization and/or buphthalmos |
| |
cT3d |
Hyphema and/or massive vitreous hemorrhage |
| |
cT3e |
Aseptic orbital cellulitis |
| cT4 |
Extraocular tumor(s) involving the orbit, including optic nerve |
| |
cT4a |
Radiologic evidence of retrobulbar optic nerve involvement or thickening of optic nerve or involvement of orbital tissues |
| |
cT4b |
Extraocular tumor clinically evident with proptosis and/or an orbital mass |
To further assess the significance of tumor seeding, a multicenter, international, registry-based analysis of eyes with retinoblastoma investigated whether the distribution and clinical characteristics of retinoblastoma seeds in cT2b eyes affect local treatment failure. Of the 624 eyes in which eye salvage was attempted, 592 had complete data for globe-salvage analysis. The distribution of seeds was focal in 143 eyes (24.2%) and diffuse in 449 eyes (75.8%). At presentation, diffuse seeding was associated with a 2.8-fold risk of eventual local treatment failure, compared with focal retinoblastoma seeding. The 5-year Kaplan-Meier cumulative globe-salvage rate (without external-beam radiation therapy) was 78% for eyes with focal seeding and 49% for eyes with diffuse seeding. This subclassification of retinoblastoma seeding is not currently included in the AJCC staging system.[3][Level of evidence C3]
Table 4. Definition of Pathological Primary Tumor (pT)a
| pT Category |
pT Criteria |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| pTX |
Unknown evidence of intraocular tumor |
| pT0 |
No evidence of intraocular tumor |
| pT1 |
Intraocular tumor(s) without any local invasion, focal choroidal invasion, or pre- or intralaminar involvement of the optic nerve head |
| pT2 |
Intraocular tumor(s) with local invasion |
| |
pT2a |
Concomitant focal choroidal invasion and pre- or intralaminar involvement of the optic nerve head |
| |
pT2b |
Tumor invasion of stroma of iris and/or trabecular meshwork and/or Schlemm's canal |
| pT3 |
Intraocular tumor(s) with significant local invasion |
| |
pT3a |
Massive choroidal invasion (>3 mm in largest diameter, or multiple foci of focal choroidal involvement totalling >3 mm, or any full-thickness choroidal involvement) |
| |
pT3b |
Retrolaminar invasion of the optic nerve head, not involving the transected end of the optic nerve |
| |
pT3c |
Any partial-thickness involvement of the sclera within the inner two thirds |
| |
pT3d |
Full-thickness invasion into the outer third of the sclera and/or invasion into or around emissary channels |
| pT4 |
Evidence of extraocular tumor: tumor at the transected end of the optic nerve, tumor in the meningeal spaces around the optic nerve, full-thickness invasion of the sclera with invasion of the episclera, adjacent adipose tissue, extraocular muscle, bone, conjunctiva, or eyelids |
Table 5. Definition of Clinical Regional Lymph Node (cN)a
| cN Category |
cN Criteria |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| cNX |
Regional lymph nodes cannot be assessed |
| cN0 |
No regional lymph node involvement |
| cN1 |
Evidence of preauricular, submandibular, and cervical lymph node involvement |
Table 6. Definition of Pathological Regional Lymph Node (pN)a
| pN Category |
pN Criteria |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| pNX |
Regional lymph node involvement cannot be assessed |
| pN0 |
No lymph node involvement |
| pN1 |
Regional lymph node involvement |
Table 7. Definition of Clinical (c) and Pathological (p) Distant Metastasis (M)a
| M Category |
M Criteria |
| CNS = central nervous system. |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| cM0 |
No signs or symptoms of intracranial or distant metastasis |
| cM1 |
Distant metastasis without microscopic confirmation |
| |
cM1a |
Tumor(s) involving any distant site (e.g., bone marrow, liver) on clinical or radiologic tests |
| |
cM1b |
Tumor involving the CNS on radiologic imaging (not including trilateral retinoblastoma) |
| pM1 |
Distant metastasis with histopathologic confirmation |
| |
pM1a |
Histopathologic confirmation of tumor at any distant site (e.g., bone marrow, liver, or other) |
| |
pM1b |
Histopathologic confirmation of tumor in the cerebrospinal fluid or CNS parenchyma |
Table 8. Histologic Grade (G)a
| G |
G Definition |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| GX |
Grade cannot be assessed |
| G1 |
Tumor with areas of retinoma (fleurettes or neuronal differentiation) |
| G2 |
Tumor with many rosettes (Flexner-Wintersteiner or Homer Wright) |
| G3 |
Tumor with occasional rosettes (Flexner-Wintersteiner or Homer Wright) |
| G4 |
Tumor with poorly differentiated cells without rosettes and/or with extensive areas (more than half of tumor) of anaplasia |
Table 9. AJCC Prognostic Stage Groups: Clinical Stage (cTNM)a
| When cT is... |
And N is... |
And M is... |
And H is... |
Then the clinical stage group is... |
| cM = clinical distant metastasis; cN = clinical regional lymph node; cT = clinical primary tumor; H = heritable trait; pM = pathological distant metastasis. |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| cT1, cT2, cT3 |
cN0 |
cM0 |
Any |
I |
| cT4a |
cN0 |
cM0 |
Any |
II |
| cT4b |
cN0 |
cM0 |
Any |
III |
| Any |
cN1 |
cM0 |
Any |
III |
| Any |
Any |
cM1 or pM1 |
Any |
IV |
Table 10. AJCC Prognostic Stage Groups: Pathological Stage (pTNM)a
| When pT is... |
And N is... |
And M is... |
And H is... |
Then the pathological stage group is... |
| cM = clinical distant metastasis; H = heritable trait; pT = pathological primary tumor; pN = pathological regional lymph node; pM = pathological distant metastasis. |
|
a Reprinted with permission from AJCC: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831. |
| pT1, pT2, pT3 |
pN0 |
cM0 |
Any |
I |
| pT4 |
pN0 |
cM0 |
Any |
II |
| Any |
pN1 |
cM0 |
Any |
III |
| Any |
Any |
cM1 or pM1 |
Any |
IV |
Size criteria
No uniform size criteria exist for intraocular retinoblastoma associated with the presence of high-risk pathological features. An international, multicenter, registry-based, retrospective case series from 13 countries was used to assess the association of high-risk pathological features at diagnosis (defined as AJCC stages pT3 and pT4) with high-risk clinical features (defined as AJCC stages cT2 and cT3) and a newly proposed AJCC Ophthalmic Oncology Task Force (OOTF) Size Grouping system. AJCC-OOTF divided intraocular tumor size into the following four groups:[4][Level of evidence C3]
This same international, multicenter, registry-based, retrospective case series was used to assess the risk of metastatic death. The analysis was based on presenting features (n = 1,814 patients with clinical cT2 or cT3 stages; n = 1,416 patients for tumor size) and treatment in patients with advanced intraocular retinoblastoma. Advanced retinoblastoma for this study was defined by AJCC categories cT2 and cT3 and AJCC-OOTF Size Groups. Treatments were primary enucleation, systemic chemotherapy with secondary enucleation, and systemic chemotherapy with eye salvage.[5][Level of evidence C3]
- Increasing cT3 subcategories were associated with a higher risk of metastatic death. There was an estimated 4.9-fold risk for cT3c, a 14.0-fold risk for cT3d, and a 19.6-fold risk for cT3e, compared with cT2a.
- Increasing age at presentation (median age of diagnosis, 22 months vs. 16 months; P < .001) and attempt at eye salvage by systemic chemotherapy were also significant risk factors for metastasis.
- Increasing intraocular Size Group was associated with an increased risk of metastatic death. There was a 10.0-fold risk for Size Group 3 tumors and a 41.1-fold risk for Size Group 4 tumors, compared with Size Group 1 tumors.
International Retinoblastoma Staging System (IRSS)
The more simplified IRSS has been proposed by an international consortium of ophthalmologists and pediatric oncologists.[6] The IRSS is more widely used in the clinical setting than the AJCC staging system (see Table 11). A retrospective German study found that the IRSS predicted survival in 633 children with heritable retinoblastoma, 582 of whom presented with IRSS stage 0 or I disease.[7]
Table 11. International Retinoblastoma Staging System
| Stage |
Description |
| CNS = central nervous system; CSF = cerebrospinal fluid. |
| 0 |
Eye has not been enucleated and no dissemination of disease. For more information, see the International Classification of Retinoblastomasection. |
| I |
Eye enucleated, completely resected histologically |
| II |
Eye enucleated, microscopic residual tumor |
| III |
Regional extension |
a. Overt orbital disease |
| b. Preauricular or cervical lymph node extension |
| IV |
Metastatic disease |
a. Hematogenous metastasis (without CNS involvement) |
| —Single lesion |
| —Multiple lesions |
| b. CNS extension (with or without any other site of regional or metastatic disease) |
| —Prechiasmatic lesion |
| —CNS mass |
| —Leptomeningeal and CSF disease |
Grouping Systems
The following grouping systems are relevant for assessment of intraocular disease extension and are helpful predictors of ocular salvage:
- International Classification of Retinoblastoma.
- American Joint Committee on Cancer (AJCC), 8th edition.
- Reese-Ellsworth classification for intraocular tumors (historical).
International Classification of Retinoblastoma
The International Classification of Retinoblastoma grouping system was developed with the goal of providing a simpler, more user-friendly classification that is more applicable to current therapies. This newer system is based on the extent of tumor seeding within the vitreous cavity and subretinal space, rather than on tumor size and location (see Table 12). The use of this system seems to better predict treatment success.[8,9,10] This system may also help predict high-risk histopathology. In a study of more than 500 patients with retinoblastoma, histopathological evidence of high-risk disease was noted in 17% of Group D eyes and 24% of Group E eyes. This can be helpful in counseling parents regarding the potential need for postoperative systemic therapy.[11]
Table 12. The International Classification of Retinoblastoma Grouping System
| Group |
Definition |
|
Group A
|
Small intraretinal tumors away from the foveola and disc. |
All tumors are 3 mm or smaller in greatest dimension, confined to the retinaand |
| All tumors are located further than 3 mm from the foveola and 1.5 mm from the optic disc. |
|
Group B
|
All remaining discrete tumors confined to the retina. |
All other tumors confined to the retina not in Group A. |
| Tumor-associated subretinal fluid less than 3 mm from the tumor with no subretinal seeding. |
| Tumor located closer than 3 mm to the optic nerve or fovea. |
|
Group C
|
Discrete local disease with minimal subretinal or vitreous seeding. |
Tumor(s) are discrete. |
| Subretinal fluid, present or past, without seeding involving up to one-fourth of the retina. |
| Local fine vitreous seeding may be present close to the discrete tumor. |
| Local subretinal seeding less than 3 mm (2 DD) from the tumor. |
|
Group D
|
Diffuse disease with significant vitreous or subretinal seeding. |
Tumor(s) may be massive or diffuse. |
| Subretinal fluid present or past without seeding, involving up to total retinal detachment. |
| Diffuse or massive vitreous disease may includegreasy seeds or avascular tumor masses. |
| Diffuse subretinal seeding may include subretinal plaques or tumor nodules. |
|
Group E
|
Presence of any one or more of the following poor prognosis features: |
Tumor touching the lens. |
| Tumor anterior to anterior vitreous face involving ciliary body or anterior segment. |
| Diffuse infiltrating retinoblastoma. |
| Neovascular glaucoma. |
| Opaque media from hemorrhage. |
| Tumor necrosis with aseptic orbital cellulites. |
| Phthisis bulbi. |
Reese-Ellsworth Classification for Intraocular Tumors
Reese and Ellsworth developed a classification system for intraocular retinoblastoma that has been shown to have prognostic significance for maintenance of sight and control of local disease at a time when surgery and external-beam radiation therapy were the primary treatment options. However, developments in the conservative management of intraocular retinoblastoma have made the Reese-Ellsworth grouping system less predictive for eye salvage and less helpful in guiding treatment.[9] This grouping system is seldom used and serves largely as a historical reference.
References:
-
Chantada GL, Sampor C, Bosaleh A, et al.: Comparison of staging systems for extraocular retinoblastoma: analysis of 533 patients. JAMA Ophthalmol 131 (9): 1127-34, 2013.
-
Mallipatna A, Gallie BL, Chevez-Barrios P, et al.: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 819-31.
-
Tomar AS, Finger PT, Gallie B, et al.: Retinoblastoma seeds: impact on American Joint Committee on Cancer clinical staging. Br J Ophthalmol 107 (1): 127-132, 2023.
-
Tomar AS, Finger PT, Gallie B, et al.: High-risk Pathologic Features Based on Presenting Findings in Advanced Intraocular Retinoblastoma: A Multicenter, International Data-Sharing American Joint Committee on Cancer Study. Ophthalmology 129 (8): 923-932, 2022.
-
Tomar AS, Finger PT, Gallie B, et al.: Metastatic Death Based on Presenting Features and Treatment for Advanced Intraocular Retinoblastoma: A Multicenter Registry-Based Study. Ophthalmology 129 (8): 933-945, 2022.
-
Chantada G, Doz F, Antoneli CB, et al.: A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer 47 (6): 801-5, 2006.
-
Temming P, Arendt M, Viehmann A, et al.: How Eye-Preserving Therapy Affects Long-Term Overall Survival in Heritable Retinoblastoma Survivors. J Clin Oncol 34 (26): 3183-8, 2016.
-
Murphree L: Staging and grouping of retinoblastoma. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 422-7.
-
Shields CL, Mashayekhi A, Au AK, et al.: The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 113 (12): 2276-80, 2006.
-
Novetsky DE, Abramson DH, Kim JW, et al.: Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet 30 (1): 40-4, 2009.
-
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
Treatment Option Overview for Retinoblastoma
Treatment planning by a multidisciplinary team of cancer specialists—including a pediatric oncologist, ophthalmologist, and radiation oncologist—with experience treating ocular tumors of childhood is required to optimize treatment outcomes.[1] Evaluation at specialized centers is highly recommended before the initiation of treatment to improve the likelihood of ocular salvage and vision preservation.
The goals of therapy include the following:
- Eradicate the disease to save the patient's life.
- Preserve as much vision as possible.
- Decrease risk of late sequelae from treatment, particularly subsequent neoplasms (SNs).
Many treatments considered to be standard of care have not been studied in a randomized fashion.
Treatment of retinoblastoma depends on the intraocular and extraocular disease burden, disease laterality, germline RB1 gene status, and the potential for preserving vision. For patients presenting with intraocular disease, particularly those with bilateral eye involvement, a conservative approach consisting of tumor reduction with intravenous or intra-arterial chemotherapy (ophthalmic artery chemotherapy), coupled with aggressive local therapy, may result in high ocular salvage rates.[2] Radiation therapy, one of the most effective treatments in retinoblastoma, is usually reserved for cases of intraocular or extraocular disease progression.
A risk-adapted, judicious combination of the following therapeutic options should be considered:
- Enucleation.
- Local treatment (cryotherapy, laser therapy, and brachytherapy).
- Systemic chemotherapy.
- Intra-arterial chemotherapy (ophthalmic artery infusion of chemotherapy).
- Intravitreal chemotherapy.
- Intracameral chemotherapy.
- Radiation therapy (external-beam radiation therapy [EBRT], brachytherapy).
The treatment options for intraocular, extraocular, and recurrent retinoblastoma are described in Table 13.
Table 13. Treatment Options for Retinoblastoma
| Treatment Group |
Treatment Options |
| CNS = central nervous system; EBRT = external-beam radiation therapy. |
| Intraocular retinoblastoma: |
|
| |
Unilateral retinoblastoma |
Enucleation for large intraocular tumors, with or without adjuvant chemotherapy |
| Conservative ocular salvage approaches when the eye and vision can be saved: |
| —Chemoreduction with either systemic or intra-arterial chemotherapy with or without intravitreal chemotherapy |
| —Local treatments (cryotherapy, thermotherapy, and plaque radiation therapy) |
| |
Bilateral retinoblastoma |
Enucleation for large intraocular tumors, followed by pathology-based, risk-adapted chemotherapy when the eye and vision cannot be saved |
| Conservative ocular salvage approaches when the eye and vision can be saved: |
| —Chemoreduction with either systemic or intra-arterial chemotherapy with or without intravitreal chemotherapy |
| —Local treatments (cryotherapy, thermotherapy, and plaque radiation therapy) |
| —EBRT |
| Extraocular retinoblastoma: |
|
| |
Orbital and locoregional retinoblastoma |
Chemotherapy |
| Radiation therapy |
| Enucleation(for extraocular extension) |
| |
CNS disease |
Systemic chemotherapy and CNS-directed therapy with radiation therapy |
| Systemic chemotherapy followed by myeloablative chemotherapy and stem cell rescue with or without radiation therapy |
| |
Synchronous trilateral retinoblastoma |
Systemic chemotherapy followed by surgery and myeloablative chemotherapy with stem cell rescue |
| Systemic chemotherapy followed by surgery and radiation therapy |
| |
Extracranial metastatic retinoblastoma |
Systemic chemotherapy followed by myeloablative chemotherapy with stem cell rescue and radiation therapy |
| Progressive or recurrent intraocular retinoblastoma |
Enucleation |
| Radiation therapy (EBRT or plaque radiation therapy) |
| Local treatments (cryotherapy or thermotherapy) |
| Salvage chemotherapy (systemic or intra-arterial) |
| Intravitreal chemotherapy, especially for refractory or recurrent vitreous seeding |
| Progressive or recurrent extraocular retinoblastoma |
Systemic chemotherapy and radiation therapy for orbital disease |
| Systemic chemotherapy followed by myeloablative chemotherapy with stem cell rescue, and radiation therapy for extraorbital disease |
Enucleation
Upfront removal of the eye is indicated for large tumors filling the vitreous for which there is little or no likelihood of restoring vision, in cases of extension to the anterior chamber, or in the presence of neovascular glaucoma. Patients must be monitored closely for orbital recurrence of disease, particularly in the first 2 years after enucleation.[3][Level of evidence C1]
Enucleation is also used as a salvage treatment in cases of disease progression or recurrence in patients receiving eye-salvage management. The pathology specimen must be carefully examined to identify patients who are at high risk of extraocular dissemination and who may require adjuvant chemotherapy.[4][Level of evidence C1 and C2]
Enucleation in patients younger than 3 years does not allow for the proper orbital growth during subsequent development, causing asymmetry of the final orbital size.[5]
Local Treatment (Cryotherapy, Laser Therapy, and Brachytherapy)
For patients undergoing eye-salvage treatments, aggressive local therapy is always required. Local treatment is administered by the ophthalmologist directly to the tumor.
-
Cryotherapy. Cryotherapy is based on the application of a cryoprobe to the sclera in the immediate vicinity of the retinal tumor. Cryotherapy is used as primary therapy or with chemotherapy for tumors smaller than 4 disc diameters (DD) in the anterior portion of the retina.
-
Laser therapy. Laser therapy may be used as primary therapy for small tumors or in combination with chemotherapy for larger tumors. Traditional photocoagulation (argon laser), in which the laser was applied around the tumor to target the tumor vasculature, has given way to thermotherapy (diode laser). Thermotherapy is delivered directly to the tumor surface via infrared wavelengths of light.[6,7]
-
Brachytherapy (plaque radiation therapy). For larger tumors that are not amenable to cryotherapy or laser therapy, brachytherapy can provide an effective means for local control. For more information, see the Radiation Therapy section.
Systemic Chemotherapy
Systemic chemotherapy plays a role in the following situations:
-
Adjuvant setting for patients with high-risk pathology. Different regimens have been used in the management of patients with high-risk pathology in the enucleated specimen. Most regimens include a three-drug combination of vincristine, etoposide, and carboplatin, either alone or alternating with cyclophosphamide and an anthracycline.[8,9,10,11]; [4][Level of evidence C1 and C2]
-
Treatment of patients with extraocular and metastatic disease. Patients with extraocular disease benefit from more intensive therapy. While a standard treatment has not been determined, responses to cisplatin-based regimens, with consolidation using high-dose chemotherapy and autologous hematopoietic stem cell rescue for patients with extraorbital disease, have been reported.[12,13,14,15]
-
Chemoreductive treatment in conjunction with aggressive local treatment for patients undergoing ocular salvage treatments. During the past two decades, the standard of care has been systemic chemotherapy to reduce tumor volume (chemoreduction) to facilitate the use of local treatments and to avoid the long-term effects of radiation therapy.[16] The success rate for eye salvage varies from center to center, but overall good ocular outcomes are consistently obtained for discrete tumors without vitreous seeding.
In a large cohort analysis of 994 eyes in 554 patients who were treated with intravenous chemotherapy and had long-term outcome data, investigators found that tumor control was strongly dependent on the International Classification of Retinoblastoma group designation per eye. Frontline intravenous chemotherapy consisting of six cycles of vincristine, etoposide, and carboplatin plus additional intra-arterial chemotherapy and/or plaque radiation therapy led to tumor control for Groups A (96%), B (91%), C (91%), D (71%), and E (32%) by year 2. With the aforementioned treatment, enucleation or external-beam radiation therapy could be avoided, and the tumor-controlling effect lasted up to 20 years.[17][Level of evidence C1]
Eye grouping, as defined by the International Classification of Retinoblastoma, is the best predictor of ocular salvage using this approach, with salvage rates ranging from 60% to 100%.[16]
Prolonged chemotherapy instead of enucleation, in the context of persistent intraocular disease activity, should be used cautiously because this approach has been associated with an increased risk of metastatic disease.[18]; [19][Level of evidence C2]
Intra-Arterial Chemotherapy (Ophthalmic Artery Infusion of Chemotherapy)
Direct delivery of chemotherapy into the eye via cannulation of the ophthalmic artery is a feasible and effective method for ocular salvage when performed at high-volume centers that have specialized services of an interventional radiologist skilled in this area and a pediatric anesthesiologist. The Children's Oncology Group conducted a multi-institutional study (ARET12P1 [NCT02097134]) to evaluate the feasibility of administering intra-arterial therapy to newly diagnosed patients with Group D retinoblastoma. The study failed to meet the feasibility goals, highlighting the importance of referring patients to high-volume institutions that have expertise in the procedure.[20] Responses to chemotherapy using this approach can be further consolidated with local control measures, as described above.
Melphalan is the most common and most effective agent used for intra-arterial chemotherapy. It is often combined with topotecan or carboplatin when responses are suboptimal or there is very advanced intraocular disease.[21,22]
Outcome after intra-arterial chemotherapy correlates with the extent of intraocular burden, as follows:
- Patients with early intraocular disease (Group B and C eyes) have an excellent outcome, with ocular salvage rates exceeding 85%. These patients may be treated with single-agent therapy.[22]
- Patients with Group D eyes have a worse outcome, with ocular salvage rates below 60%.[22] However, ocular salvage rates higher than 80% have been reported in specialized centers.[21,23] For patients with very advanced intraocular disease, an alternative treatment is the use of systemic chemotherapy followed by consolidation with intra-arterial melphalan.[24]
- Ocular salvage rates when intra-arterial chemotherapy administration is used as salvage treatment for patients with recurrent or progressive disease are consistently lower, with globe survival rates of 50% to 75%.[21,22,23] Best results are reported using a more intensive three-drug regimen with melphalan, topotecan, and carboplatin.[25]
The role of intra-arterial chemotherapy in ocular salvage has been further clarified in a multicenter randomized clinical trial. This trial compared intra-arterial chemotherapy with systemic chemotherapy for children with unilateral advanced (Group D or E) retinoblastoma. Patients were randomly assigned to receive either four cycles of intra-arterial melphalan combination chemotherapy (two cycles with carboplatin and two cycles with topotecan) or six cycles of systemic chemotherapy with vincristine, carboplatin, and etoposide. Local control was performed based on standard practice. The 2-year progression-free ocular salvage rates were 53% for patients in the intra-arterial chemotherapy group and 27% for patients in the intravenous chemotherapy group. The ocular salvage rates were 71% for patients who received intra-arterial chemotherapy and 51% for patients who received intravenous chemotherapy.[26]
Patients with bilateral disease can undergo tandem intra-arterial chemotherapy administration.[27] In those circumstances, patients are at higher risk of systemic toxicity caused by melphalan exposure,[28] and single-agent carboplatin may be used to treat the less-advanced eye during the tandem procedure.[29] For neonates and very young infants in whom the cannulization of the ophthalmic artery is not feasible, bridge treatment with single-agent systemic carboplatin until the infant is aged 3 months or weighs 6 kg, followed by consolidation with intra-arterial chemotherapy, has been shown to be very effective. In one study, the 1-year radiation-free ocular survival rate was 95%.[30]
In a study of 39 infants younger than 3 months with advanced intraocular retinoblastoma (Group D and E eyes), patients received intra-arterial chemotherapy as primary treatment (29 eyes) or secondary treatment (13 eyes previously treated with intravenous chemotherapy) using a microcatheterization procedure. The middle meningeal artery was used when the ophthalmic artery could not be catheterized.[31]
- The globe salvage rate was 96% for the 23 Group D eyes and 33% for the 6 Group E eyes, but the 2-year overall ocular survival rate was 81% because of an increased incidence of phthisis bulbi.
- The incidence of complete vision loss was high.
- Follow-up was limited, ranging from 6 months to 6 years; therefore, later effects are unknown.
The addition of intravitreal chemotherapy to intra-arterial chemotherapy appears to markedly improve the overall effectiveness in eyes with vitreous seeds, especially those with vitreous seed clouds.[21,32,33] For more information, see the Intravitreal Chemotherapy section.
In patients presenting with total retinal detachment, ophthalmic artery chemosurgery has been shown to promote retinal reattachment.[34]
Complications related to intra-arterial chemotherapy include the following:[22,26,35]
- Retinal detachment (up to 19.3%).[36]
- Vitreous hemorrhage (up to 18.1%).
- Ptosis (13.6%).
- Dysmotility (6.5%).
- Vascular and ischemic effects, including ophthalmic artery stenosis (18%) and occlusion (up to 9%).
- Optic atrophy (3.4%).
- Phthisis (2.7%).
Major vascular complications related to the procedure are very rare. Strokes or significant acute neurological events have not been reported by the most experienced groups.[21,22,37] However, stenosis of the ophthalmic artery and occlusion of the retinal artery have been documented.[35,37] The risk of thrombosis is significantly increased in children with thrombophilia.[38] In a large series of 196 patients who were treated with 682 infusions of intra-arterial chemotherapy, ophthalmic vascular events were reported in 17% of the treated eyes.[35]
The impact of the intraocular vascular changes on vision has not been fully assessed because of the young age of the first cohorts of patients treated. Most patients do not have substantial electroretinographic changes,[39] and preservation of central vision has been reported.[40] A proportion of patients with abnormal electroretinograms (ERGs) with or without retinal detachment may have improved ERGs in the years after intra-arterial chemotherapy.[41] However, in patients with heavily pretreated eyes, intensive intra-arterial chemotherapy may result in worsening of retinal function.[25]
Another risk associated with intra-arterial chemotherapy is the exposure to ionizing radiation during fluoroscopy. Mean total radiation doses of 42.3 mGy have been reported in very experienced centers.[42] After multiple procedures, cumulative doses can reach 0.1 to 0.2 Gy, which can be cataractogenic and potentially carcinogenic in this susceptible population.[43] There has been no increase in the incidence of second malignancies.[44,45] However, longer follow-up is required to fully ascertain the risks associated with the procedure.
The risk of metastatic progression with direct ocular delivery of chemotherapy appears to be very low.[2] However, up to 20 cases of patients treated with intra-arterial chemotherapy who subsequently developed metastases have been reported.[22]
Intravitreal Chemotherapy
Studies suggest that direct intravitreal injection of melphalan or topotecan may be effective in controlling active vitreous seeds.[46,47]; [48,49][Level of evidence C2] A retrospective study of 264 eyes (250 children) treated with intravitreal melphalan for vitreous seeds over a two-decade period reported a complete remission rate of 68%. There was a low incidence of extraocular spread as a result of the injection that occurred in children with high-risk features.[50][Level of evidence C2]
Because of initial concerns about the potential for tumor dissemination, the use of intravitreal chemotherapy was limited. However, additional reports have estimated that the proportion of patients with extraocular tumor spread, as the result of intravitreal injection, is negligible.[51,52] While this procedure is safe and well tolerated, recent studies have shown a direct correlation between the number of injections and a decrease in retinal function, as measured by ERG.[52]; [53][Level of evidence C3]
Preliminary data support that intra-arterial chemotherapy plus intravitreal chemotherapy (as needed for vitreous seeding) may improve globe salvage in eyes with advanced retinoblastoma when compared with children who were treated in earlier years with intra-arterial chemotherapy alone.[52]; [32][Level of evidence C2] Compared with the children treated in the earlier era, children treated in the later era received a combination of intra-arterial and intra-vitreal chemotherapy, which demonstrated shorter time to regression, fewer recurrences, fewer enucleations, and no increased toxicity, including no difference in loss of retinal function as measured by ERG.[33][Level of evidence C3]
As experience with the use of intra-vitreal chemotherapy expands, studies have demonstrated its efficacy in controlling subretinal seeds and recurrent retinal tumors, suggesting a potential role beyond the control of vitreous seeds as an adjunctive therapy in the globe-sparing treatment of retinoblastoma.[54]
Intracameral Chemotherapy
A retrospective, single-institution study reported on the treatment of anterior chamber seeding with the injection of aqueous melphalan. Ocular salvage was achieved in 6 of 11 eyes (median, four injections), with a mean follow-up of 17 months.[55]
Radiation Therapy
Retinoblastoma is a very radiosensitive malignancy.
-
EBRT. EBRT doses ranging from 35 Gy to 46 Gy usually result in long-term remissions. Because of the need to sedate young children and the intricacies of field planning, special expertise in pediatric radiation therapy is important. Radiation therapy is used in cases of progression after conservative approaches, in patients with extraocular extension, and as part of the management of patients with metastatic disease.
Newer methods of delivering EBRT are being applied to reduce adverse long-term effects. This includes intensity-modulated radiation therapy and proton-beam radiation therapy (charged-particle radiation therapy).[56,57,58,59] Preliminary data suggest that proton radiation therapy is associated with a lower risk of radiation-induced malignancy in survivors of heritable retinoblastoma.
In a nonrandomized study that compared two contemporary cohorts of patients with heritable retinoblastoma who were treated with either photon or proton radiation therapy, the 10-year cumulative incidence of radiation-induced SNs was significantly different between the two groups (0% for proton radiation vs. 14% for photon radiation, P = .015).[60]
EBRT in infants causes growth failure of the orbital bones and results in cosmetic deformity. EBRT also increases the risk of SNs in children with heritable retinoblastoma.
-
Brachytherapy (plaque radiation therapy). Indications for plaque radiation therapy include solitary tumors with a diameter ranging between 6 mm and 15 mm, tumor thickness of 10 mm or less, and tumor location of more than 3 mm from the optic disc or fovea. The most commonly used radioisotope is iodine I 125, although others such as iridium Ir 192 and ruthenium Ru 106 are also effective. In combination with the appropriate use of chemotherapy and other forms of focal consolidation, brachytherapy can be very effective in the treatment of localized retinal tumors that are not amenable to other means of local therapy.[61,62,63]
References:
-
Chintagumpala M, Chevez-Barrios P, Paysse EA, et al.: Retinoblastoma: review of current management. Oncologist 12 (10): 1237-46, 2007.
-
Abramson DH, Fabius AW, Issa R, et al.: Advanced Unilateral Retinoblastoma: The Impact of Ophthalmic Artery Chemosurgery on Enucleation Rate and Patient Survival at MSKCC. PLoS One 10 (12): e0145436, 2015.
-
Kim JW, Kathpalia V, Dunkel IJ, et al.: Orbital recurrence of retinoblastoma following enucleation. Br J Ophthalmol 93 (4): 463-7, 2009.
-
Chévez-Barrios P, Eagle RC, Krailo M, et al.: Study of Unilateral Retinoblastoma With and Without Histopathologic High-Risk Features and the Role of Adjuvant Chemotherapy: A Children's Oncology Group Study. J Clin Oncol 37 (31): 2883-2891, 2019.
-
Oatts JT, Robbins JA, de Alba Campomanes AG: The effect of enucleation on orbital growth in patients with retinoblastoma. J AAPOS 21 (4): 309-312, 2017.
-
Shields CL, Santos MC, Diniz W, et al.: Thermotherapy for retinoblastoma. Arch Ophthalmol 117 (7): 885-93, 1999.
-
Francis JH, Abramson DH, Brodie SE, et al.: Indocyanine green enhanced transpupillary thermotherapy in combination with ophthalmic artery chemosurgery for retinoblastoma. Br J Ophthalmol 97 (2): 164-8, 2013.
-
Chantada GL, Fandiño AC, Guitter MR, et al.: Results of a prospective study for the treatment of unilateral retinoblastoma. Pediatr Blood Cancer 55 (1): 60-6, 2010.
-
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
-
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
-
Sullivan EM, Wilson MW, Billups CA, et al.: Pathologic risk-based adjuvant chemotherapy for unilateral retinoblastoma following enucleation. J Pediatr Hematol Oncol 36 (6): e335-40, 2014.
-
Dunkel IJ, Khakoo Y, Kernan NA, et al.: Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer 55 (1): 55-9, 2010.
-
Matsubara H, Makimoto A, Higa T, et al.: A multidisciplinary treatment strategy that includes high-dose chemotherapy for metastatic retinoblastoma without CNS involvement. Bone Marrow Transplant 35 (8): 763-6, 2005.
-
Rodriguez-Galindo C, Wilson MW, Haik BG, et al.: Treatment of metastatic retinoblastoma. Ophthalmology 110 (6): 1237-40, 2003.
-
Kremens B, Wieland R, Reinhard H, et al.: High-dose chemotherapy with autologous stem cell rescue in children with retinoblastoma. Bone Marrow Transplant 31 (4): 281-4, 2003.
-
Rodriguez-Galindo C, Orbach DB, VanderVeen D: Retinoblastoma. Pediatr Clin North Am 62 (1): 201-23, 2015.
-
Shields CL, Bas Z, Tadepalli S, et al.: Long-term (20-year) real-world outcomes of intravenous chemotherapy (chemoreduction) for retinoblastoma in 964 eyes of 554 patients at a single centre. Br J Ophthalmol 104 (11): 1548-1555, 2020.
-
Zhao J, Dimaras H, Massey C, et al.: Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J Clin Oncol 29 (7): 845-51, 2011.
-
Berry JL, Kogachi K, Aziz HA, et al.: Risk of metastasis and orbital recurrence in advanced retinoblastoma eyes treated with systemic chemoreduction versus primary enucleation. Pediatr Blood Cancer 64 (4): , 2017.
-
Chintagumpala M, Piao J, Gombos D, et al.: A multi-institutional feasibility study of intra-arterial chemotherapy in children with retinoblastoma. A Children's Oncology Group study (COG ARET12P1). Pediatr Blood Cancer 71 (1): e30718, 2024.
-
Francis JH, Levin AM, Zabor EC, et al.: Ten-year experience with ophthalmic artery chemosurgery: Ocular and recurrence-free survival. PLoS One 13 (5): e0197081, 2018.
-
Yousef YA, Soliman SE, Astudillo PPP, et al.: Intra-arterial Chemotherapy for Retinoblastoma: A Systematic Review. JAMA Ophthalmol 134 (5): 584-591, 2016.
-
Abramson DH, Daniels AB, Marr BP, et al.: Intra-Arterial Chemotherapy (Ophthalmic Artery Chemosurgery) for Group D Retinoblastoma. PLoS One 11 (1): e0146582, 2016.
-
Shields CL, Kaliki S, Al-Dahmash S, et al.: Management of advanced retinoblastoma with intravenous chemotherapy then intra-arterial chemotherapy as alternative to enucleation. Retina 33 (10): 2103-9, 2013 Nov-Dec.
-
Marr BP, Brodie SE, Dunkel IJ, et al.: Three-drug intra-arterial chemotherapy using simultaneous carboplatin, topotecan and melphalan for intraocular retinoblastoma: preliminary results. Br J Ophthalmol 96 (10): 1300-3, 2012.
-
Wen X, Fan J, Jin M, et al.: Intravenous versus super-selected intra-arterial chemotherapy in children with advanced unilateral retinoblastoma: an open-label, multicentre, randomised trial. Lancet Child Adolesc Health 7 (9): 613-620, 2023.
-
Abramson DH, Marr BP, Francis JH, et al.: Simultaneous Bilateral Ophthalmic Artery Chemosurgery for Bilateral Retinoblastoma (Tandem Therapy). PLoS One 11 (6): e0156806, 2016.
-
Schaiquevich P, Buitrago E, Taich P, et al.: Pharmacokinetic analysis of melphalan after superselective ophthalmic artery infusion in preclinical models and retinoblastoma patients. Invest Ophthalmol Vis Sci 53 (7): 4205-12, 2012.
-
Francis JH, Gobin YP, Brodie SE, et al.: Experience of intra-arterial chemosurgery with single agent carboplatin for retinoblastoma. Br J Ophthalmol 96 (9): 1270-1, 2012.
-
Gobin YP, Dunkel IJ, Marr BP, et al.: Combined, sequential intravenous and intra-arterial chemotherapy (bridge chemotherapy) for young infants with retinoblastoma. PLoS One 7 (9): e44322, 2012.
-
Chen Q, Zhang B, Dong Y, et al.: Intra-arterial chemotherapy as primary or secondary treatment for infants diagnosed with advanced retinoblastoma before 3 months of age. BMC Cancer 19 (1): 693, 2019.
-
Shields CL, Alset AE, Say EA, et al.: Retinoblastoma Control With Primary Intra-arterial Chemotherapy: Outcomes Before and During the Intravitreal Chemotherapy Era. J Pediatr Ophthalmol Strabismus 53 (5): 275-84, 2016.
-
Francis JH, Iyer S, Gobin YP, et al.: Retinoblastoma Vitreous Seed Clouds (Class 3): A Comparison of Treatment with Ophthalmic Artery Chemosurgery with or without Intravitreous and Periocular Chemotherapy. Ophthalmology 124 (10): 1548-1555, 2017.
-
Rowlands MA, Mondesire-Crump I, Levin A, et al.: Total retinal detachments due to retinoblastoma: Outcomes following intra-arterial chemotherapy/ophthalmic artery chemosurgery. PLoS One 13 (4): e0195395, 2018.
-
Ancona-Lezama D, Dalvin LA, Lucio-Alvarez JA, et al.: OPHTHALMIC VASCULAR EVENTS AFTER INTRA-ARTERIAL CHEMOTHERAPY FOR RETINOBLASTOMA: Real-World Comparison Between Primary and Secondary Treatments. Retina 39 (12): 2264-2272, 2019.
-
Shields CL, Say EAT, Pefkianaki M, et al.: RHEGMATOGENOUS RETINAL DETACHMENT AFTER INTRAARTERIAL CHEMOTHERAPY FOR RETINOBLASTOMA: The 2016 Founders Award Lecture. Retina 37 (8): 1441-1450, 2017.
-
Shields CL, Bianciotto CG, Jabbour P, et al.: Intra-arterial chemotherapy for retinoblastoma: report No. 2, treatment complications. Arch Ophthalmol 129 (11): 1407-15, 2011.
-
Francis JH, Gobin YP, Nagiel A, et al.: Thrombophilia in patients with retinoblastoma receiving ophthalmic artery chemosurgery. Arch Ophthalmol 130 (12): 1605-8, 2012.
-
Abramson DH: Chemosurgery for retinoblastoma: what we know after 5 years. Arch Ophthalmol 129 (11): 1492-4, 2011.
-
Brodie SE, Munier FL, Francis JH, et al.: Persistence of retinal function after intravitreal melphalan injection for retinoblastoma. Doc Ophthalmol 126 (1): 79-84, 2013.
-
Abdelhakim AH, Francis JH, Marr BP, et al.: Retinal reattachment and ERG recovery after ophthalmic artery chemosurgery for advanced retinoblastoma in eyes with minimal baseline retinal function. Br J Ophthalmol 101 (5): 623-628, 2017.
-
Boddu SR, Abramson DH, Marr BP, et al.: Selective ophthalmic artery chemosurgery (SOAC) for retinoblastoma: fluoroscopic time and radiation dose parameters. A baseline study. J Neurointerv Surg 9 (11): 1107-1112, 2017.
-
Vijayakrishnan R, Shields CL, Ramasubramanian A, et al.: Irradiation toxic effects during intra-arterial chemotherapy for retinoblastoma: should we be concerned? Arch Ophthalmol 128 (11): 1427-31, 2010.
-
Suzuki S, Yamane T, Mohri M, et al.: Selective ophthalmic arterial injection therapy for intraocular retinoblastoma: the long-term prognosis. Ophthalmology 118 (10): 2081-7, 2011.
-
Habib LA, Francis JH, Fabius AW, et al.: Second primary malignancies in retinoblastoma patients treated with intra-arterial chemotherapy: the first 10 years. Br J Ophthalmol 102 (2): 272-275, 2018.
-
Francis JH, Abramson DH, Gaillard MC, et al.: The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology 122 (6): 1173-9, 2015.
-
Shields CL, Manjandavida FP, Arepalli S, et al.: Intravitreal melphalan for persistent or recurrent retinoblastoma vitreous seeds: preliminary results. JAMA Ophthalmol 132 (3): 319-25, 2014.
-
Ghassemi F, Shields CL: Intravitreal melphalan for refractory or recurrent vitreous seeding from retinoblastoma. Arch Ophthalmol 130 (10): 1268-71, 2012.
-
Munier FL, Gaillard MC, Balmer A, et al.: Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol 96 (8): 1078-83, 2012.
-
Suzuki S, Aihara Y, Fujiwara M, et al.: Intravitreal injection of melphalan for intraocular retinoblastoma. Jpn J Ophthalmol 59 (3): 164-72, 2015.
-
Smith SJ, Smith BD: Evaluating the risk of extraocular tumour spread following intravitreal injection therapy for retinoblastoma: a systematic review. Br J Ophthalmol 97 (10): 1231-6, 2013.
-
Francis JH, Brodie SE, Marr B, et al.: Efficacy and Toxicity of Intravitreous Chemotherapy for Retinoblastoma: Four-Year Experience. Ophthalmology 124 (4): 488-495, 2017.
-
Smith SJ, Smith BD, Mohney BG: Ocular side effects following intravitreal injection therapy for retinoblastoma: a systematic review. Br J Ophthalmol 98 (3): 292-7, 2014.
-
Abramson DH, Ji X, Francis JH, et al.: Intravitreal chemotherapy in retinoblastoma: expanded use beyond intravitreal seeds. Br J Ophthalmol 103 (4): 488-493, 2019.
-
Munier FL, Moulin A, Gaillard MC, et al.: Intracameral Chemotherapy for Globe Salvage in Retinoblastoma with Secondary Anterior Chamber Invasion. Ophthalmology 125 (4): 615-617, 2018.
-
Krasin MJ, Crawford BT, Zhu Y, et al.: Intensity-modulated radiation therapy for children with intraocular retinoblastoma: potential sparing of the bony orbit. Clin Oncol (R Coll Radiol) 16 (3): 215-22, 2004.
-
Reisner ML, Viégas CM, Grazziotin RZ, et al.: Retinoblastoma--comparative analysis of external radiotherapy techniques, including an IMRT technique. Int J Radiat Oncol Biol Phys 67 (3): 933-41, 2007.
-
Lee CT, Bilton SD, Famiglietti RM, et al.: Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys 63 (2): 362-72, 2005.
-
Mouw KW, Sethi RV, Yeap BY, et al.: Proton radiation therapy for the treatment of retinoblastoma. Int J Radiat Oncol Biol Phys 90 (4): 863-9, 2014.
-
Sethi RV, Shih HA, Yeap BY, et al.: Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer 120 (1): 126-33, 2014.
-
Shields CL, Shields JA, Cater J, et al.: Plaque radiotherapy for retinoblastoma: long-term tumor control and treatment complications in 208 tumors. Ophthalmology 108 (11): 2116-21, 2001.
-
Merchant TE, Gould CJ, Wilson MW, et al.: Episcleral plaque brachytherapy for retinoblastoma. Pediatr Blood Cancer 43 (2): 134-9, 2004.
-
Shields CL, Mashayekhi A, Sun H, et al.: Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology 113 (11): 2087-92, 2006.
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence has been slowly increasing since 1975.[1] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following pediatric specialists and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Transplant surgeons.
- Pathologists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists and hematologists.
- Ophthalmologists.
- Rehabilitation specialists.
- Pediatric oncology nurses.
- Social workers.
- Child-life professionals.
- Psychologists.
- Nutritionists.
For specific information about supportive care for children and adolescents with cancer, see the summaries on Supportive and Palliative Care.
The American Academy of Pediatrics has outlined guidelines for pediatric cancer centers and their role in the treatment of children and adolescents with cancer.[2] At these centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate is offered to most patients and their families. Clinical trials for children and adolescents diagnosed with cancer are generally designed to compare potentially better therapy with current standard therapy. Other types of clinical trials test novel therapies when there is no standard therapy for a cancer diagnosis. Most of the progress in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[3,4,5] Between 1975 and 2020, childhood cancer mortality decreased by more than 50%.[3,6,7] Childhood and adolescent cancer survivors require close monitoring because side effects of cancer therapy may persist or develop months or years after treatment. For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
-
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
-
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed December 15, 2023.
-
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
-
Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.
-
Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.
-
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 15, 2023.
-
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed March 6, 2024.
Treatment of Intraocular Retinoblastoma
Treatment of Unilateral Intraocular Retinoblastoma
Treatment options for unilateral intraocular retinoblastoma include the following:
- Enucleation for large intraocular tumors, with or without adjuvant chemotherapy based on pathology risk, when the eye cannot be saved.
- Conservative ocular salvage approaches when the eye and vision can be saved:
- Chemoreduction with either systemic or intra-arterial chemotherapy with or without intravitreal chemotherapy.
- Local treatments, including cryotherapy, thermotherapy, and plaque radiation therapy.
Enucleation with or without adjuvant chemotherapy
Because unilateral disease is usually massive and there is often no expectation that useful vision can be preserved, up-front surgery (enucleation) is commonly performed. Careful examination of the enucleated specimen by an experienced pathologist is necessary to determine whether high-risk features for metastatic disease are present. These high-risk features include the following:[1,2,3,4,5]; [6][Level of evidence C1 and C2]
- Anterior chamber seeding.
- Massive choroidal involvement.
- Tumor beyond the lamina cribrosa.
- Scleral and extrascleral extension.
Pre-enucleation magnetic resonance imaging has low sensitivity and specificity for the detection of high-risk pathology.[7]
High-risk pathology has been associated with the presence of minimal dissemination in bone marrow and cerebrospinal fluid using quantitative polymerase chain reaction for detection of CRX or GD2 synthase. In a group of 96 children with nonmetastatic retinoblastoma and high-risk pathology, the 3-year disease-free survival rate was 78% for patients with detectable minimal dissemination, compared with 98% for those without detectable disease (P = .004).[8]
Systemic adjuvant therapy with vincristine, doxorubicin, and cyclophosphamide or with vincristine, carboplatin, and etoposide has been used to prevent the development of metastatic disease in patients with certain high-risk features assessed by pathological review after enucleation.[3]
The Children's Oncology Group ARET0332 (NCT00335738) trial prospectively studied the role of adjuvant chemotherapy in 321 eligible children with newly diagnosed enucleated unilateral retinoblastoma. Central histopathological review was performed for all patients' pathology slides. Defined indications for adjuvant chemotherapy included massive choroid replacement defined as posterior uveal invasion grades IIC and IID, any posterior uveal involvement less than 3 mm with concomitant optic nerve involvement, and optic nerve involvement posterior to the lamina cribrosa. Treatment consisted of six cycles of carboplatin, etoposide, and vincristine administered every 4 weeks.[6][Level of evidence C1 and C2]
- Central review of enucleated eyes by ophthalmic pathologists identified a group of children (19% of all eligible patients) who were misclassified by the institutional pathology report.
- The importance of universal guidelines for handling and interpreting retinoblastoma specimens is instrumental in the recognition and categorization of high-risk features.
- For all patients on the study, the overall 2-year event-free survival (EFS) rate was 98%, and the overall survival (OS) rate was 99%.
- For patients with high-risk features requiring adjuvant chemotherapy, the 2-year EFS rate was 96%.
- For patients with low-risk features for whom observation was indicated, the 2-year EFS rate was 99%.
- The combination of extensive involvement of both the choroid and the optic nerve posterior to the lamina cribrosa represents the highest risk of recurrence and supports the need for more intensive chemotherapy for this group.
- The average time to first recurrence was less than 12 months, and the time to death after recurrence was approximately 8 months.
Conservative ocular salvage approaches
Conservative ocular salvage approaches, such as systemic chemotherapy and local-control treatments, may be offered in an attempt to save the eye and preserve vision.[9] Ocular salvage rates correlate with intraocular grouping. The possibility of saving the eye without the use of external-beam radiation therapy (EBRT) exceeds 80% for children with early intraocular disease. However, the ocular outcomes for children with advanced intraocular disease are poor using systemic chemotherapy and local treatments, with less than 40% ocular salvage rates, even after the use of EBRT.[10] Plaque brachytherapy has been used as a salvage therapy for patients with unilateral retinoblastoma. In one series, 12 eyes in 12 children were treated with ruthenium plaque brachytherapy. Globe salvage was achieved in 75% of the patients. The ultimate local control rate was 66%.[11]
Caution must be used when delaying enucleation by extending treatment with systemic chemotherapy when tumor control does not appear to be possible, particularly for Group E eyes. Pre-enucleation chemotherapy for eyes with advanced intraocular disease may result in downstaging and underestimate the pathological evidence of extraretinal and extraocular disease, thus increasing the risk of dissemination.[12]
The delivery of chemotherapy via ophthalmic artery cannulation as initial treatment for advanced unilateral retinoblastoma appears to be more effective than systemic chemotherapy for chemoreduction, particularly for Group D eyes.[13,14]; [15][Level of evidence C3] In a multidisciplinary state-of-the-art center, intra-arterial chemotherapy to treat patients with advanced intraocular unilateral retinoblastoma may result in ocular salvage rates of approximately 70% to 90%.[14,15,16,17,18] For more information, see the Intra-Arterial Chemotherapy (Ophthalmic Artery Infusion of Chemotherapy) section.
Electroretinography, a technique that measures the electrical responses of various cell types in the retina, including the photoreceptors, can be used to assess retinal function during and after treatment with intra-arterial chemotherapy. In one study, pretreatment electroretinography correlated with final visual acuity after treatment with intra-arterial chemotherapy, suggesting that this technique may potentially be used to help with treatment strategy decisions and prioritization of interventions.[19]
Treatment options under clinical evaluation for unilateral intraocular retinoblastoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
-
ARET2121 (NCT05504291) (A Study to Give Treatment Inside the Eye to Treat Retinoblastoma): This trial is exploring the feasibility of incorporating intravitreal melphalan injections during neoadjuvant systemic chemotherapy (carboplatin, vincristine, and etoposide). Patients are eligible if they have unilateral Group D retinoblastoma with vitreous seeding or bilateral retinoblastoma with the worst eye categorized as Group D and vitreous seeding.
Follow-up after treatment of unilateral intraocular retinoblastoma
Because a proportion of children who present with unilateral retinoblastoma will eventually develop disease in the opposite eye, these children undergo genetic counseling and testing and periodic examinations of the unaffected eye, regardless of the treatment they receive. Asynchronous bilateral disease occurs most frequently in patients with affected parents and in children diagnosed during the first months of life.
Treatment of Bilateral Intraocular Retinoblastoma
The goal of therapy for bilateral retinoblastoma is ocular and vision preservation and the delay or avoidance of EBRT and enucleation.
Treatment options for bilateral intraocular retinoblastoma include the following:
- Enucleation for large intraocular tumors, followed by pathology-based, risk-adapted chemotherapy when the eye and vision cannot be saved.
- Conservative ocular salvage approaches when the eye and vision can be saved:
- Chemoreduction with either systemic or intra-arterial chemotherapy with or without intravitreal chemotherapy.
- Local treatments, including cryotherapy, thermotherapy, and plaque radiation therapy.
- EBRT.
Intraocular tumor burden is usually asymmetrical, and treatment is dictated by the most advanced eye. Systemic therapy is generally selected based on the eye with more extensive disease. Treatment options described for unilateral disease may be applied to one or both affected eyes in patients with bilateral disease. While up-front enucleation of an advanced eye and risk-adapted adjuvant chemotherapy may be required, a more conservative approach using primary chemoreduction and aggressive local treatments with close monitoring for response is usually the treatment of choice. EBRT is now reserved for patients whose eyes do not respond adequately to primary systemic or intra-arterial chemotherapy and local consolidation.[20]
Several large centers have published trial results that used systemic chemotherapy in conjunction with aggressive local consolidation for patients with bilateral disease.[21] The backbone of chemoreduction has generally been carboplatin, etoposide, and vincristine. While the less toxic combination of vincristine and carboplatin can provide disease control for a significant proportion of patients, ocular salvage appears to be superior when etoposide is included in the regimen.[22][Level of evidence B1] A single-institution study achieved similar results when topotecan was substituted for etoposide in a combination regimen.[23][Level of evidence C1] Using chemoreduction and aggressive local control consolidation, the International Classification of Retinoblastoma grouping system has been proven to predict ocular survival, with globe salvage rates usually exceeding 80% for Groups A and B, and 40% to 80% for Groups C and D, although EBRT may be required in more advanced intraocular cases.[24]; [21,23][Level of evidence C1]
Delivery of chemotherapy via ophthalmic artery cannulation with the addition of intra-vitreal chemotherapy for patients with persistent vitreous or subretinal disease has become a very strong alternative to the use of systemic chemotherapy.[13,14,16,17]; [15][Level of evidence C3] While tandem administration is feasible, bilateral administrations increase the risk of systemic toxicity caused by melphalan exposure.[25] In these circumstances, intra-arterial chemotherapy with single-agent carboplatin may be used to treat the less-advanced eye during the tandem procedure.[26] These treatments should only be performed in an experienced center with a state-of-the-art treatment infrastructure and a dedicated multidisciplinary team. For more information, see the sections on Intra-Arterial Chemotherapy (Ophthalmic Artery Infusion of Chemotherapy) and Intravitreal Chemotherapy.
For patients with large intraocular tumor burdens with subretinal or vitreous seeds (Group D eyes), the administration of higher doses of carboplatin coupled with subtenon carboplatin, and the addition of lower doses of EBRT (36 Gy) for patients with persistent disease has been explored. Using this intensive approach, eye survival may approach a rate of 70% at 60 months.[27][Level of evidence B4]
The prognosis for patients with Group E eyes who are treated with systemic chemotherapy and local control measures is very poor without radiation therapy.[27][Level of evidence B4] The use of prolonged systemic chemotherapy for Group E eyes to avoid or delay enucleation has been associated with lower disease-specific survival.[12][Level of evidence C1]
Treatment options under clinical evaluation for bilateral intraocular retinoblastoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
-
ARET2121 (NCT05504291) (A Study to Give Treatment Inside the Eye to Treat Retinoblastoma): This trial is exploring the feasibility of incorporating intravitreal melphalan injections during neoadjuvant systemic chemotherapy (carboplatin, vincristine, and etoposide). Patients are eligible if they have unilateral Group D retinoblastoma with vitreous seeding or bilateral retinoblastoma with the worst eye categorized as Group D and vitreous seeding.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Chantada GL, Guitter MR, Fandiño AC, et al.: Treatment results in patients with retinoblastoma and invasion to the cut end of the optic nerve. Pediatr Blood Cancer 52 (2): 218-22, 2009.
-
Eagle RC: High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med 133 (8): 1203-9, 2009.
-
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
-
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
-
Sastre X, Chantada GL, Doz F, et al.: Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med 133 (8): 1199-202, 2009.
-
Chévez-Barrios P, Eagle RC, Krailo M, et al.: Study of Unilateral Retinoblastoma With and Without Histopathologic High-Risk Features and the Role of Adjuvant Chemotherapy: A Children's Oncology Group Study. J Clin Oncol 37 (31): 2883-2891, 2019.
-
Chawla B, Sharma S, Sen S, et al.: Correlation between clinical features, magnetic resonance imaging, and histopathologic findings in retinoblastoma: a prospective study. Ophthalmology 119 (4): 850-6, 2012.
-
Laurent VE, Torbidoni AV, Sampor C, et al.: Minimal Disseminated Disease in Nonmetastatic Retinoblastoma With High-Risk Pathologic Features and Association With Disease-Free Survival. JAMA Ophthalmol 134 (12): 1374-1379, 2016.
-
Shields CL, Honavar SG, Meadows AT, et al.: Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radiotherapy or enucleation. Am J Ophthalmol 133 (5): 657-64, 2002.
-
Shields CL, Honavar SG, Meadows AT, et al.: Chemoreduction for unilateral retinoblastoma. Arch Ophthalmol 120 (12): 1653-8, 2002.
-
Negretti GS, Quhill H, Duncan C, et al.: Ruthenium plaque radiotherapy in the current era of retinoblastoma treatment. Ophthalmic Genet 43 (6): 756-761, 2022.
-
Zhao J, Dimaras H, Massey C, et al.: Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J Clin Oncol 29 (7): 845-51, 2011.
-
Abramson DH, Fabius AW, Issa R, et al.: Advanced Unilateral Retinoblastoma: The Impact of Ophthalmic Artery Chemosurgery on Enucleation Rate and Patient Survival at MSKCC. PLoS One 10 (12): e0145436, 2015.
-
Munier FL, Mosimann P, Puccinelli F, et al.: First-line intra-arterial versus intravenous chemotherapy in unilateral sporadic group D retinoblastoma: evidence of better visual outcomes, ocular survival and shorter time to success with intra-arterial delivery from retrospective review of 20 years of treatment. Br J Ophthalmol 101 (8): 1086-1093, 2017.
-
Shields CL, Jorge R, Say EA, et al.: Unilateral Retinoblastoma Managed With Intravenous Chemotherapy Versus Intra-Arterial Chemotherapy. Outcomes Based on the International Classification of Retinoblastoma. Asia Pac J Ophthalmol (Phila) 5 (2): 97-103, 2016 Mar-Apr.
-
Francis JH, Iyer S, Gobin YP, et al.: Retinoblastoma Vitreous Seed Clouds (Class 3): A Comparison of Treatment with Ophthalmic Artery Chemosurgery with or without Intravitreous and Periocular Chemotherapy. Ophthalmology 124 (10): 1548-1555, 2017.
-
Francis JH, Levin AM, Zabor EC, et al.: Ten-year experience with ophthalmic artery chemosurgery: Ocular and recurrence-free survival. PLoS One 13 (5): e0197081, 2018.
-
Wen X, Fan J, Jin M, et al.: Intravenous versus super-selected intra-arterial chemotherapy in children with advanced unilateral retinoblastoma: an open-label, multicentre, randomised trial. Lancet Child Adolesc Health 7 (9): 613-620, 2023.
-
Levin AM, Francis JH, McFadden M, et al.: Association of electroretinography with visual outcomes after ophthalmic artery chemosurgery for retinoblastoma in ICRb D and E eyes. PLoS One 14 (1): e0210647, 2019.
-
Orman A, Koru-Sengul T, Miao F, et al.: The modern role of radiation therapy in treating advanced-stage retinoblastoma: long-term outcomes and racial differences. Int J Radiat Oncol Biol Phys 90 (5): 1037-43, 2014.
-
Rodriguez-Galindo C, Orbach DB, VanderVeen D: Retinoblastoma. Pediatr Clin North Am 62 (1): 201-23, 2015.
-
Lumbroso-Le Rouic L, Aerts I, Hajage D, et al.: Conservative treatment of retinoblastoma: a prospective phase II randomized trial of neoadjuvant chemotherapy followed by local treatments and chemothermotherapy. Eye (Lond) 30 (1): 46-52, 2016.
-
Brennan RC, Qaddoumi I, Mao S, et al.: Ocular Salvage and Vision Preservation Using a Topotecan-Based Regimen for Advanced Intraocular Retinoblastoma. J Clin Oncol 35 (1): 72-77, 2017.
-
Shields CL, Mashayekhi A, Au AK, et al.: The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 113 (12): 2276-80, 2006.
-
Schaiquevich P, Buitrago E, Taich P, et al.: Pharmacokinetic analysis of melphalan after superselective ophthalmic artery infusion in preclinical models and retinoblastoma patients. Invest Ophthalmol Vis Sci 53 (7): 4205-12, 2012.
-
Francis JH, Gobin YP, Brodie SE, et al.: Experience of intra-arterial chemosurgery with single agent carboplatin for retinoblastoma. Br J Ophthalmol 96 (9): 1270-1, 2012.
-
Berry JL, Jubran R, Kim JW, et al.: Long-term outcomes of Group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRT salvage. Pediatr Blood Cancer 60 (4): 688-93, 2013.
Treatment of Extraocular Retinoblastoma
In high-income countries, few patients with retinoblastoma present with extraocular disease. Extraocular disease may be localized to the soft tissues surrounding the eye or to the optic nerve beyond the margin of resection. However, further extension may progress into the brain and meninges, with subsequent seeding of the spinal fluid and as distant metastatic disease involving the lungs, bones, and bone marrow.
Treatment of Orbital and Locoregional Retinoblastoma
Orbital retinoblastoma occurs as a result of tumor progression through the emissary vessels and sclera. For this reason, transscleral disease is considered to be extraocular and should be treated as such. Orbital retinoblastoma is isolated in 60% to 70% of cases.
Treatment options for extraocular retinoblastoma (orbital and locoregional) include the following:
- Chemotherapy.
- Radiation therapy.
- Enucleation (for extraocular extension).
Treatment includes systemic chemotherapy and radiation therapy. With this treatment approach, 60% to 85% of patients can be cured. Because most recurrences occur in the central nervous system (CNS), regimens that include drugs with well-documented CNS penetration are used.
The Children's Oncology Group (COG) performed a prospective international trial (ARET0321 [NCT00554788]) that included patients with extraocular retinoblastoma. The study showed that intensified therapy improved the outcomes of patients with stage II, III or IVa disease, compared with historical controls. However, stage IVb patients need more effective therapy.[1][Level of evidence B4]
- Patients with stage II or III retinoblastoma (n = 19) received four cycles of chemotherapy (vincristine, cisplatin, cyclophosphamide, etoposide) followed by involved-field radiation therapy (45 Gy).
- The median follow-up was 7.3 years.
- The 3-year event-free survival (EFS) and overall survival (OS) rates were 88.1%.
- Patients with stage IVa retinoblastoma (n = 18) received four cycles of induction therapy (noted above). Patients with more than a partial response underwent autologous hematopoietic stem cell support. Patients with residual tumor after chemotherapy received radiation therapy.
- The 3-year EFS and OS rates were 76.7%.
- Patients with stage IVb retinoblastoma (n = 20) received the same treatment as patients with stage IVa disease.
- The 1-year EFS rate was 28.3%, and the OS rate was 42.1%.
- The 3-year EFS rate was 14.2%, and the OS rate was 12.3%.
- Six patients developed secondary malignancies. There was one case each of osteosarcoma (8 years after treatment), thyroid papillary carcinoma (9 years after treatment), glioblastoma multiforme (8 years after treatment), large B-cell Epstein-Barr virus–associated lymphoproliferative disorder (8 months after treatment), acute myeloid leukemia (2 years after treatment), and alveolar soft part sarcoma of the bladder (5 years after treatment). Five patients had unilateral retinoblastoma, two of whom received radiation therapy (one each with unilateral and bilateral).
For patients with macroscopic orbital disease, delay of surgery until response to chemotherapy is achieved (usually after receiving two or three courses of treatment) has been effective. Patients then undergo enucleation and receive an additional four to six courses of chemotherapy. During consolidation, the patient receives local control therapies with orbital irradiation (40–45 Gy). Using this approach, orbital exenteration is not indicated.[2]
Patients with isolated involvement of the optic nerve at the transsection level are considered to have extraocular disease and are treated using systemic therapy, similar to that used for macroscopic orbital disease, and irradiation of the entire orbit (36 Gy) with a 10 Gy boost to the chiasm (total of 46 Gy).[3]
Treatment of CNS Disease
Intracranial dissemination occurs by direct extension through the optic nerve. The prognosis for these patients is dismal. Treatment includes platinum-based, intensive systemic chemotherapy and CNS-directed therapy. Although intrathecal chemotherapy has been used traditionally, there is no preclinical or clinical evidence to support its use.
Treatment options for extraocular retinoblastoma (CNS disease) include the following:
- Systemic chemotherapy and CNS-directed therapy with radiation therapy.
- Systemic chemotherapy followed by myeloablative chemotherapy and stem cell rescue with or without radiation therapy.
The administration of radiation therapy to these patients is controversial. Responses have been observed with craniospinal radiation using 25 Gy to 35 Gy to the entire craniospinal axis and a boost (10 Gy) to sites of measurable disease.[1]
The COG conducted a prospective study (ARET0321 [NCT00554788]) of patients with extraocular retinoblastoma, which included patients with stage IVb disease who were treated with four cycles of induction therapy (vincristine, cisplatin, cyclophosphamide, and etoposide).[1][Level of evidence B4]
- Patients who had more than a partial response were then treated with high-dose carboplatin, thiotepa, and etoposide with autologous hematopoietic stem cell support.
- Patients with residual disease after chemotherapy received radiation therapy.
- In the 20 patients with stage IVb disease, the 1-year EFS rate was 28.3%, and the OS rate was 42.1%. The 3-year EFS rate was 14.2%, and the OS rate was 12.3%.
Treatment of Synchronous Trilateral Retinoblastoma
Trilateral retinoblastoma is usually associated with a pineal lesion or, less commonly, a suprasellar lesion.[4,5,6] In patients with the heritable form of retinoblastoma, CNS disease is less likely the result of metastatic or regional spread than of a primary intracranial focus, such as a pineal tumor. The prognosis for patients with trilateral retinoblastoma is very poor. Most patients die of disseminated neuraxis disease in less than 9 months.[7,8] However, with increased surveillance and aggressive therapy, there has been improvement in survival, from 6% (patients treated before 1995) to 44% (patients treated after 1996).[9]
Treatment options for synchronous trilateral retinoblastoma include the following:
- Systemic chemotherapy followed by surgery and myeloablative chemotherapy with stem cell rescue.
- Systemic chemotherapy followed by surgery and radiation therapy.
While pineoblastomas occurring in older patients are sensitive to radiation therapy, current strategies are directed towards avoiding radiation by using intensive chemotherapy followed by consolidation with myeloablative chemotherapy and autologous hematopoietic progenitor cell rescue. This approach is similar to those being used in the treatment of brain tumors in infants.[10]
For more information about trilateral retinoblastoma, including screening with neuroimaging, see the Trilateral retinoblastoma section.
Treatment of Extracranial Metastatic Retinoblastoma
Treatment options for extracranial metastatic retinoblastoma include the following:
- Systemic chemotherapy followed by myeloablative chemotherapy with stem cell rescue and radiation therapy.
Hematogenous metastases may develop in the bones, bone marrow, and, less frequently, the liver. The COG conducted a prospective international trial (ARET0321 [NCT00554788]) for patients with extraocular retinoblastoma. The study showed that intensified therapy improved the outcomes of patients with stage IVa disease.[1][Level of evidence B4] Patients with stage IVa retinoblastoma (n = 18) were treated with four cycles of induction therapy (vincristine, cisplatin, cyclophosphamide, and etoposide).
- The patients who had more than a partial response then received one cycle of high-dose carboplatin, thiotepa, and etoposide with autologous hematopoietic stem cell support. Patients with residual tumor after chemotherapy received radiation therapy.
- With a median follow up of 7.3 years, the 3-year EFS and OS rates were 76.7%.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Dunkel IJ, Piao J, Chantada GL, et al.: Intensive Multimodality Therapy for Extraocular Retinoblastoma: A Children's Oncology Group Trial (ARET0321). J Clin Oncol 40 (33): 3839-3847, 2022.
-
Radhakrishnan V, Kashyap S, Pushker N, et al.: Outcome, pathologic findings, and compliance in orbital retinoblastoma (International Retinoblastoma Staging System stage III) treated with neoadjuvant chemotherapy: a prospective study. Ophthalmology 119 (7): 1470-7, 2012.
-
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
-
Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
-
Paulino AC: Trilateral retinoblastoma: is the location of the intracranial tumor important? Cancer 86 (1): 135-41, 1999.
-
Blach LE, McCormick B, Abramson DH, et al.: Trilateral retinoblastoma--incidence and outcome: a decade of experience. Int J Radiat Oncol Biol Phys 29 (4): 729-33, 1994.
-
Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
-
Marcus DM, Brooks SE, Leff G, et al.: Trilateral retinoblastoma: insights into histogenesis and management. Surv Ophthalmol 43 (1): 59-70, 1998 Jul-Aug.
-
de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
-
Dunkel IJ, Jubran RF, Gururangan S, et al.: Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer 54 (3): 384-7, 2010.
Treatment of Progressive or Recurrent Retinoblastoma
The prognosis for a patient with progressive or recurrent retinoblastoma depends on the site and extent of the progression or recurrence and previous treatment received.
The introduction of intravenous chemotherapy for the treatment of retinoblastoma in the early 1990s revolutionized retinoblastoma management. In a retrospective review of 869 eyes in 551 patients with retinoblastoma who were treated with chemoreduction, 64% of the eyes experienced a recurrence and 94% of the recurrences or new tumors were detected within the first 3 years of treatment. Risk factors for recurrence included the following:[1][Level of evidence C1]
- Younger patient age at diagnosis (odds ratio [OR], 1.02 per 1 month decrease; P = .02).
- More advanced International Classification of Retinoblastoma group (OR, 1.24 per 1 more advanced group; P = .01).
- Closer tumor distance to the optic disc (OR, 1.11 per 1 mm decrease; P = .03).
- Presentation with subretinal seeds (OR, 1.66; P = .02).
- Presence of germline pathogenic variants, which contributed to the risk of new tumor formation.
Intraocular and extraocular recurrences have very different prognoses and are treated in different ways.
Treatment of Progressive or Recurrent Intraocular Retinoblastoma
Treatment options for progressive or recurrent intraocular retinoblastoma include the following:
- Enucleation.
- Radiation therapy (external-beam or plaque radiation therapy).
- Local treatments (cryotherapy or thermotherapy).
- Salvage chemotherapy (systemic or intra-arterial chemotherapy).
- Intravitreal chemotherapy, especially for refractory or recurrent vitreous seeding.
For more information about the use and potential applications of intra-arterial and intravitreal chemotherapy, see the sections on Intra-Arterial Chemotherapy (Ophthalmic Artery Infusion of Chemotherapy) and Intravitreal Chemotherapy.
New intraocular tumors can arise in patients with the heritable form of disease whose eyes have been treated with local control measures only because every cell in the retina carries the RB1 variant. This event should not be considered a recurrence. Even with previous treatment consisting of chemoreduction and local control measures in very young patients with heritable retinoblastoma, surveillance may detect new tumors at an early stage. Additional local control therapy, including plaque radiation therapy, can successfully eradicate these tumors.[2]
When the recurrence or progression of retinoblastoma is confined to the eye and is small, the prognosis for sight and survival may be excellent with local therapy only.[3][Level of evidence C3] If the recurrence or progression is confined to the eye but is extensive, the prognosis for sight is poor; however, survival remains excellent.
Intra-arterial chemotherapy (IAC) into the ophthalmic artery has been effective in patients who relapse after systemic chemotherapy and radiation therapy.[4,5] Rescue IAC, usually with other agents, has also been used after primary IAC.[6] Plaque radiation therapy is an option for patients who have a retinoblastoma recurrence after IAC treatment.[7][Level of evidence C3] Radiation therapy should be considered for patients who have not been previously irradiated. Finally, enucleation may be required in cases of progressive disease after all eye-salvaging treatments have failed.
Treatment of Progressive or Recurrent Extraocular Retinoblastoma
Treatment options for progressive or recurrent extraocular retinoblastoma include the following:
- Systemic chemotherapy and radiation therapy for orbital disease.
- Systemic chemotherapy followed by myeloablative chemotherapy with stem cell rescue and radiation therapy for extraorbital disease.
Recurrence in the orbit after enucleation is treated with aggressive chemotherapy in addition to local radiation therapy because of the high risk of metastatic disease.[8][Level of evidence C1] After enucleation for recurrence, high-resolution magnetic resonance imaging with orbital coils can be helpful in distinguishing orbital recurrence from postsurgical enhancement.[9]
If the recurrence or progression is extraocular, the chance of survival is poor.[10] However, the use of intensive systemic chemotherapy and consolidation with high-dose chemotherapy and autologous hematopoietic stem cell rescue may improve the chance of a cure, particularly for patients with extracranial recurrence. For patients with disease recurrence after those intensive approaches, clinical trials may be considered. For more information, see the Treatment of Extraocular Retinoblastoma section.
Treatment Options Under Clinical Evaluation for Progressive or Recurrent Retinoblastoma
One approach under investigation for patients with progressive intraocular retinoblastoma includes the use of an oncolytic adenovirus that targets RB1.[11]
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References:
-
Dalvin LA, Bas Z, Tadepalli S, et al.: Risk Factors for Tumor Recurrence Following Primary Intravenous Chemotherapy (Chemoreduction) for Retinoblastoma in 869 Eyes of 551 Patients. J Pediatr Ophthalmol Strabismus 57 (4): 224-234, 2020.
-
Wilson MW, Haik BG, Billups CA, et al.: Incidence of new tumor formation in patients with hereditary retinoblastoma treated with primary systemic chemotherapy: is there a preventive effect? Ophthalmology 114 (11): 2077-82, 2007.
-
Chan MP, Hungerford JL, Kingston JE, et al.: Salvage external beam radiotherapy after failed primary chemotherapy for bilateral retinoblastoma: rate of eye and vision preservation. Br J Ophthalmol 93 (7): 891-4, 2009.
-
Schaiquevich P, Ceciliano A, Millan N, et al.: Intra-arterial chemotherapy is more effective than sequential periocular and intravenous chemotherapy as salvage treatment for relapsed retinoblastoma. Pediatr Blood Cancer 60 (5): 766-70, 2013.
-
Say EA, Iyer PG, Hasanreisoglu M, et al.: Secondary and tertiary intra-arterial chemotherapy for massive persistent or recurrent subretinal retinoblastoma seeds following previous chemotherapy exposure: long-term tumor control and globe salvage in 30 eyes. J AAPOS 20 (4): 337-42, 2016.
-
Francis JH, Abramson DH, Gobin YP, et al.: Efficacy and toxicity of second-course ophthalmic artery chemosurgery for retinoblastoma. Ophthalmology 122 (5): 1016-22, 2015.
-
Ruben M, Eiger-Moscovich M, Yaghy A, et al.: Iodine-125 Plaque Radiotherapy for Retinoblastoma Recurrence Following Intra-arterial Chemotherapy. J Pediatr Ophthalmol Strabismus 59 (3): 164-171, 2022 May-Jun.
-
Kim JW, Kathpalia V, Dunkel IJ, et al.: Orbital recurrence of retinoblastoma following enucleation. Br J Ophthalmol 93 (4): 463-7, 2009.
-
Sirin S, de Jong MC, de Graaf P, et al.: High-Resolution Magnetic Resonance Imaging Can Reliably Detect Orbital Tumor Recurrence after Enucleation in Children with Retinoblastoma. Ophthalmology 123 (3): 635-45, 2016.
-
Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
-
Pascual-Pasto G, Bazan-Peregrino M, Olaciregui NG, et al.: Therapeutic targeting of the RB1 pathway in retinoblastoma with the oncolytic adenovirus VCN-01. Sci Transl Med 11 (476): , 2019.
Late Effects of Retinoblastoma Therapy
In a report from the Retinoblastoma Survivor Study (N = 470), 87% of survivors of retinoblastoma (mean age, 43 years; median follow-up, 42 years) had at least one medical condition and 71% had a severe or life-threatening condition. Compared with patients without retinoblastoma, the adjusted relative risk of a chronic condition in survivors was 1.4 (P < .01). The relative risk of a grade 3 or 4 condition was 7.6 (P < .01). After excluding ocular conditions and subsequent neoplasms (SNs), this excess risk was found to persist only for patients with bilateral disease.[1]
Subsequent Neoplasms (SNs)
SNs are the most common cause of death in patients with retinoblastoma. SNs contribute to about 50% of deaths in patients with both bilateral disease and genetically defined heritable retinoblastoma.[2,3,4] Survivors of retinoblastoma have a high risk of developing SNs.
Factors that influence the risk of SNs include the following:
-
Heritable retinoblastoma. Patients with heritable retinoblastoma have a markedly increased incidence of SNs, independent of treatment with radiation therapy.[2,3,5] In a German series of 643 patients with heritable retinoblastoma, chemotherapy with or without radiation therapy was the only significant risk factor for the onset of second cancers outside the periorbital region.[3] A possible association between the type of RB1 variant and incidence of SNs may exist, with complete loss of RB1 activity associated with a higher incidence of SNs.[6] Patients who are heterozygous for regular-penetrance RB1 variants may be at a higher risk of developing SNs than patients with incomplete-penetrance RB1 variants.[7] Regular penetrance refers to patients with heritable disease and classic multifocal bilateral tumors. Incomplete penetrance, while rare, is a clinical phenotype in which affected family members may not develop retinoblastoma or they present with unilateral disease. With the increase in survival of patients with heritable retinoblastoma, it has become apparent that they are also at risk of developing epithelial cancers late in adulthood. A marked increase in mortality from lung, bladder, and other epithelial cancers has been described.[8,9]
In a large series from two institutions, 2,053 patients with retinoblastoma (diagnosed between 1914–2016) were identified, with a maximum of 70 years of follow-up. Most deaths occurred in patients with hereditary retinoblastoma (518 of 1,129), and 267 of these deaths were caused by SNs. Increased risk of death resulting from cancers of the pancreas, large intestines, and kidney were reported. Overall risk of SNs was greater for patients who were treated with radiation therapy and chemotherapy compared with patients who were treated with radiation therapy alone, although patterns varied by organ site. In a cohort of 143 retinoblastoma survivors diagnosed between 1997 and 2006, continued improvements in mortality were seen.[4][Level of evidence C1] For patients with nonhereditary retinoblastoma, only 27 deaths in 924 patients were attributed to SNs.
Among retinoblastoma survivors with heritable retinoblastoma, those with an inherited germline pathogenic variant are at a slightly higher risk of developing an SN than are those with a de novo variant. Melanoma was the most common SN seen in patients with germline pathogenic variants.[10]
-
Past treatment for retinoblastoma with radiation therapy. The cumulative incidence of SNs was reported to be 26% (± 10%) in nonirradiated patients and 58% (± 10%) in irradiated patients by 50 years after diagnosis of retinoblastoma, resulting in a rate of about 1% per year.[11]
A German series of 633 patients with heritable retinoblastoma demonstrated a 5-year survival rate of 93%. However, 40 years later, only 80% of patients survived, with most succumbing to radiation-induced SNs (hazard ratio, approximately 3).[12] Other studies analyzing cohorts of patients treated with more advanced radiation planning and delivery technology have reported the SN rates to be about 9.4% in nonirradiated patients and about 30.4% in irradiated patients.[13]
In a nonrandomized study that compared two contemporary cohorts of patients with hereditary retinoblastoma who were treated with either photon (n = 31) or proton (n = 55) therapy, the 10-year cumulative incidence of radiation-induced SNs was significantly different between the two groups (0% for proton radiation vs. 14% for photon radiation; P = .015).[14] Longer follow-up is required to further define the risk of SNs associated with proton radiation.
The most common SN is sarcoma, specifically osteosarcoma, followed by soft tissue sarcoma and melanoma. These malignancies may occur inside or outside of the radiation field, although most are radiation induced. The carcinogenic effect of radiation therapy is associated with the dose delivered, particularly for subsequent sarcomas. A step-wise increase is apparent at all dose categories. In irradiated patients, two-thirds of SNs occur within irradiated tissue, and one-third of SNs occur outside the radiation field.[5,11,13,15]
In a cohort of 952 irradiated survivors of hereditary retinoblastoma who were originally diagnosed between 1914 and 2006, 105 bone sarcomas and 125 soft tissue sarcomas were identified. Approximately two-thirds of these cancers occurred in the head and neck. The incidence rates were 2,000-fold higher for bone sarcomas and 500-fold higher for soft tissue sarcomas than was expected in the general population. Head and neck bone and soft tissue sarcomas were diagnosed in early childhood and continued into adulthood, with a 60-year cumulative incidence of 6.8% for bone sarcomas and 9.3% for soft tissue sarcomas. Bone and soft tissue sarcomas diagnosed elsewhere in the body were increased 169-fold and 45.7-fold, respectively, compared with the general population. Bone sarcomas primarily occurred in the long bones during adolescence. The incidence of soft tissue sarcomas was rare until age 30 years, when it rose steeply (60-year cumulative incidence, 6.6%), particularly for females (9.4%). The soft tissue sarcomas that occurred in females were leiomyosarcomas and were mainly located in the abdomen and pelvis.[5,11]; [16][Level of evidence C1]
-
Age at time of radiation therapy. The risk of SNs also appears to depend on the patient's age at the time that external-beam radiation therapy (EBRT) is administered, especially in children younger than 12 months. The histopathological types of SNs may be influenced by age.[13,17,18]
-
Previous SN. Patients who survive SNs are at a sevenfold increased risk of developing another SN.[19] The risk increases an additional threefold for patients treated with radiation therapy.[20]
The issue of balancing long-term tumor control with the consequences of chemotherapy is unresolved. Most patients who receive chemotherapy are exposed to etoposide, which has been associated with secondary leukemia in patients without a predisposition to cancer. However, most patients are exposed at modest rates when compared with the risks associated with EBRT in heritable retinoblastoma.
Despite the known increased risk of acute myeloid leukemia (AML) associated with the use of etoposide, patients with heritable retinoblastoma are not at an increased risk of developing this SN.[21,22,23] An initial report conducted by informal survey methods described 15 patients who developed AML after chemotherapy. One-half of the patients also received radiation therapy.[22] This finding has not been substantiated by formal studies. In a single-institution study of 245 patients who received etoposide, only 1 patient developed acute promyelocytic leukemia after 79 months.[21] Additionally, the Surveillance, Epidemiology, and End Results (SEER) Program calculated standardized incidence rates for secondary hematopoietic malignancies in 34,867 survivors of childhood cancer. The observed-to-expected ratio of secondary AML in patients treated for retinoblastoma was zero.[24]
Survival from SNs is certainly suboptimal and varies widely across studies.[8,25,26,27,28,29] However, with advances in therapy, it is essential that all SNs in survivors of retinoblastoma be treated with curative intent.[30]
Other Late Effects
Other late effects that may occur after treatment for retinoblastoma include the following:
-
Diminished orbital growth. Orbital growth is somewhat diminished after enucleation. However, the impact of enucleation on orbital volume may be minimized after placement of an orbital implant.[31] Asymmetry of orbital size that develops after enucleation is greater in younger children and infants, but enucleation after age 3 years does not result in orbital asymmetry.[32]
-
Visual-field deficits. Patients with retinoblastoma demonstrate a variety of long-term visual-field defects after treatment for their intraocular disease. These defects are related to tumor size, location, and treatment method.[33]
One study of visual acuity after treatment with systemic chemotherapy and local ophthalmic therapy was conducted in 54 eyes of 40 children. After a mean follow-up of 68 months, 27 eyes (50%) had a final visual acuity of 20/40 or better, and 36 eyes (67%) had a final visual acuity of 20/200 or better. The clinical factors that predicted visual acuity of 20/40 or better were a tumor margin of at least 3 mm from the foveola and optic disc and an absence of subretinal fluid.[34]
-
Strabismus. In a study of 42 survivors of retinoblastoma (64 eyes) who underwent successful conservative treatment, 69% of patients were noted to have strabismus at final follow-up (mean, 93.7 months). Exotropia was the most common type of strabismus reported. Conservative treatments included intravenous chemotherapy (62 eyes), intra-arterial chemotherapy (10 eyes), brachytherapy (11 eyes), transpupillary thermotherapy (22 eyes), cryotherapy (47 eyes), and external-beam radiation therapy (4 eyes). On multivariate analysis, only foveal involvement was found to be a significant risk factor for developing strabismus (P < .001).[35]
-
Hearing loss. Because systemic carboplatin is now commonly used in the treatment of retinoblastoma, concern has been raised about hearing loss related to therapy. For more information about treatment, see the sections on Treatment of Intraocular Retinoblastoma and Treatment of Extraocular Retinoblastoma.
While two large studies that included children treated with six cycles of carboplatin-containing therapy (18.6 mg/kg per cycle) showed an incidence of treatment-related hearing loss of lower than 1%,[36,37] a separate series documented some degree of hearing loss in 17% of patients.[38] In the latter study, younger age (younger than 6 months at the time of treatment) and higher carboplatin systemic exposures correlated with an increased risk of ototoxicity.[38,39]
-
Neurocognitive functioning. Early studies of intellectual functioning in survivors of retinoblastoma suggested above average intelligence among survivors of bilateral disease compared with unaffected siblings and the general population, especially in those who were blind as a result of their disease.[40,41,42]
Later studies have yielded mixed results with conflicting findings, in part, resulting from the low test-retest reliability of measures used to assess cognitive outcomes at a very young age, as well as temporal differences in treatment exposures.
- Serial assessment of cognitive and adaptive functioning in a group of survivors younger than 6 years revealed declines in developmental functioning over time, with the most pronounced declines observed in patients with 13q deletions.[43]
- Further longitudinal follow-up of this cohort identified improvement in adaptive functioning in all treatment groups and improvement in cognitive functioning in survivors who were treated with enucleation alone between the ages of 5 years and 10 years.[44]
- At age 10 years, overall functioning was generally within the average range, although estimated intelligence quotient was significantly below the normative mean for children who were treated with enucleation alone.[44]
- A study of very long-term adult survivors of retinoblastoma, who were on average 33 years postdiagnosis, demonstrated largely average cognitive functioning across domains of intelligence, memory, attention, and executive function.[45]
For specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors, see Late Effects of Treatment for Childhood Cancer.
References:
-
Friedman DN, Chou JF, Oeffinger KC, et al.: Chronic medical conditions in adult survivors of retinoblastoma: Results of the Retinoblastoma Survivor Study. Cancer 122 (5): 773-81, 2016.
-
Shinohara ET, DeWees T, Perkins SM: Subsequent malignancies and their effect on survival in patients with retinoblastoma. Pediatr Blood Cancer 61 (1): 116-9, 2014.
-
Temming P, Arendt M, Viehmann A, et al.: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer 64 (1): 71-80, 2017.
-
Kleinerman RA, Tucker MA, Sigel BS, et al.: Patterns of Cause-Specific Mortality Among 2053 Survivors of Retinoblastoma, 1914-2016. J Natl Cancer Inst 111 (9): 961-969, 2019.
-
MacCarthy A, Bayne AM, Brownbill PA, et al.: Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer 108 (12): 2455-63, 2013.
-
Dommering CJ, Marees T, van der Hout AH, et al.: RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer 11 (2): 225-33, 2012.
-
Ketteler P, Hülsenbeck I, Frank M, et al.: The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma. Eur J Cancer 133: 47-55, 2020.
-
Fletcher O, Easton D, Anderson K, et al.: Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst 96 (5): 357-63, 2004.
-
Marees T, van Leeuwen FE, de Boer MR, et al.: Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer 45 (18): 3245-53, 2009.
-
Kleinerman RA, Yu CL, Little MP, et al.: Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol 30 (9): 950-7, 2012.
-
Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
-
Temming P, Arendt M, Viehmann A, et al.: How Eye-Preserving Therapy Affects Long-Term Overall Survival in Heritable Retinoblastoma Survivors. J Clin Oncol 34 (26): 3183-8, 2016.
-
Kleinerman RA, Tucker MA, Tarone RE, et al.: Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol 23 (10): 2272-9, 2005.
-
Sethi RV, Shih HA, Yeap BY, et al.: Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer 120 (1): 126-33, 2014.
-
Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007.
-
Kleinerman RA, Schonfeld SJ, Sigel BS, et al.: Bone and Soft-Tissue Sarcoma Risk in Long-Term Survivors of Hereditary Retinoblastoma Treated With Radiation. J Clin Oncol 37 (35): 3436-3445, 2019.
-
Abramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology 105 (4): 573-9; discussion 579-80, 1998.
-
Moll AC, Imhof SM, Schouten-Van Meeteren AY, et al.: Second primary tumors in hereditary retinoblastoma: a register-based study, 1945-1997: is there an age effect on radiation-related risk? Ophthalmology 108 (6): 1109-14, 2001.
-
Abramson DH, Melson MR, Dunkel IJ, et al.: Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmology 108 (10): 1868-76, 2001.
-
Marees T, van Leeuwen FE, Schaapveld M, et al.: Risk of third malignancies and death after a second malignancy in retinoblastoma survivors. Eur J Cancer 46 (11): 2052-8, 2010.
-
Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.
-
Gombos DS, Hungerford J, Abramson DH, et al.: Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology 114 (7): 1378-83, 2007.
-
Tamboli D, Topham A, Singh N, et al.: Retinoblastoma: A SEER Dataset Evaluation for Treatment Patterns, Survival, and Second Malignant Neoplasms. Am J Ophthalmol 160 (5): 953-8, 2015.
-
Rihani R, Bazzeh F, Faqih N, et al.: Secondary hematopoietic malignancies in survivors of childhood cancer: an analysis of 111 cases from the Surveillance, Epidemiology, and End Result-9 registry. Cancer 116 (18): 4385-94, 2010.
-
Yu CL, Tucker MA, Abramson DH, et al.: Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst 101 (8): 581-91, 2009.
-
Aerts I, Pacquement H, Doz F, et al.: Outcome of second malignancies after retinoblastoma: a retrospective analysis of 25 patients treated at the Institut Curie. Eur J Cancer 40 (10): 1522-9, 2004.
-
Eng C, Li FP, Abramson DH, et al.: Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst 85 (14): 1121-8, 1993.
-
Dunkel IJ, Gerald WL, Rosenfield NS, et al.: Outcome of patients with a history of bilateral retinoblastoma treated for a second malignancy: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 30 (1): 59-62, 1998.
-
Marees T, Moll AC, Imhof SM, et al.: Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst 100 (24): 1771-9, 2008.
-
Moll AC, Imhof SM, Bouter LM, et al.: Second primary tumors in patients with retinoblastoma. A review of the literature. Ophthalmic Genet 18 (1): 27-34, 1997.
-
Chojniak MM, Chojniak R, Testa ML, et al.: Abnormal orbital growth in children submitted to enucleation for retinoblastoma treatment. J Pediatr Hematol Oncol 34 (3): e102-5, 2012.
-
Oatts JT, Robbins JA, de Alba Campomanes AG: The effect of enucleation on orbital growth in patients with retinoblastoma. J AAPOS 21 (4): 309-312, 2017.
-
Abramson DH, Melson MR, Servodidio C: Visual fields in retinoblastoma survivors. Arch Ophthalmol 122 (9): 1324-30, 2004.
-
Demirci H, Shields CL, Meadows AT, et al.: Long-term visual outcome following chemoreduction for retinoblastoma. Arch Ophthalmol 123 (11): 1525-30, 2005.
-
Fabian ID, Stacey AW, Naeem Z, et al.: Strabismus in retinoblastoma survivors with long-term follow-up. J AAPOS 22 (4): 276.e1-276.e7, 2018.
-
Lambert MP, Shields C, Meadows AT: A retrospective review of hearing in children with retinoblastoma treated with carboplatin-based chemotherapy. Pediatr Blood Cancer 50 (2): 223-6, 2008.
-
Batra A, Thakar A, Bakhshi S: Ototoxicity in retinoblastoma survivors treated with carboplatin based chemotherapy: A cross-sectional study of 116 patients. Pediatr Blood Cancer 62 (11): 2060, 2015.
-
Qaddoumi I, Bass JK, Wu J, et al.: Carboplatin-associated ototoxicity in children with retinoblastoma. J Clin Oncol 30 (10): 1034-41, 2012.
-
Leahey A: A cautionary tale: dosing chemotherapy in infants with retinoblastoma. J Clin Oncol 30 (10): 1023-4, 2012.
-
Levitt EA, Rosenbaum AL, Willerman L, et al.: Intelligence of retinoblastoma patients and their siblings. Child Dev 43 (3): 939-48, 1972.
-
Eldridge R, O'Meara K, Kitchin D: Superior intelligence in sighted retinoblastoma patients and their families. J Med Genet 9 (3): 331-5, 1972.
-
Williams M: Superior intelligence of children blinded from retinoblastoma. Arch Dis Child 43 (228): 204-10, 1968.
-
Willard VW, Qaddoumi I, Chen S, et al.: Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol 32 (25): 2788-93, 2014.
-
Willard VW, Qaddoumi I, Pan H, et al.: Cognitive and Adaptive Functioning in Youth With Retinoblastoma: A Longitudinal Investigation Through 10 Years of Age. J Clin Oncol 39 (24): 2676-2684, 2021.
-
Brinkman TM, Merchant TE, Li Z, et al.: Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer 121 (1): 123-31, 2015.
Latest Updates to This Summary (07 / 24 / 2024)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
This summary was reformatted.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® Cancer Information for Health Professionals pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of retinoblastoma. It is intended as a resource to inform and assist clinicians in the care of their patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Retinoblastoma Treatment are:
- Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- D. Williams Parsons, MD, PhD (Texas Children's Hospital)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
- Nita Louise Seibel, MD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as "NCI's PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary]."
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Retinoblastoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/retinoblastoma/hp/retinoblastoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389442]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either "standard" or "under clinical evaluation." These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website's Email Us.
Last Revised: 2024-07-24